La bioquímica es el estudio de las moléculas y las reacciones químicas de la vida. Es la disciplina que emplea los principios y el lenguaje de la química a fin de explicar la biología a nivel molecular. Estudia los elementos que forman parte de la naturaleza de los seres vivos. Dentro de los elementos podemos mencionar que existen seis elementos no metálicos oxígeno, carbono, hidrógeno, nitrógeno, fósforo y azufre que representan más de 97% del peso de la mayoría de los organismos. Todos estos elementos pueden formar enlaces covalentes estables.
Los tipos de compuestos orgánicos que se encuentran comúnmente en la bioquímica los cuales son: alcohol, aldehído, cetona, acido carboxílico, tiol (sulfhidrilo) y aminas (primaria, secundaria y terciarias).
Las reacciones bioquímicas incluyen uniones químicas específicas o partes de moléculas denominadas grupos funcionales, estos son: hidroxilo, acido carbonilo, carboxilato, amino fosfato, fosforilo.
Algunos tipos de enlaces presentes en los derivados de compuestos son: éster, éter, amida, éster fosfato, fosfoanhidrido.
v Muchas macromoléculas importantes son polímeros: Las macromoléculas biológicas forman un polímero creado mediante la unión de muchas moléculas orgánicas más pequeñas, o monómeros, por medio de condensaciones (la remoción de los elementos de lagua). Cada monómero incorporado a una cadena macromolecular se denomina residuo. Las macromoléculas se clasifican en: Proteínas, polisacáridos, Ácidos nucleicos y lípidos.
v Células

Es la unidad básica de la vida. Todos los organismos son unicelulares o están compuestos por muchas células. Las células existen en una variedad extraordinaria de tamaños y formas, pero todas se pueden clasificar cómo: eucarióticas o procarióticas. Las células procariotas son células sin núcleo celular definido, es decir, cuyo material genético se encuentra disperso en el citoplasma. Las células eucariotas son células con un núcleo celular delimitado dentro de una doble capa lipídica.
Agua





 Los valores de pKa de los ácidos débiles se determinan por titulación.
Los valores de pKa de los ácidos débiles se determinan por titulación.
Ejemplo se titula una solución de ácido acético agregando pequeñas alícuotas de una base fuerte de concentración conocida. Se mide el pH de la solución y se grafica en función de la cantidad de equivalentes molares de base fuerte agregados durante la titulación. Cuando se ha titulado el ácido con la mitad de un equivalente de base, la concentración del ácido acético no disociado es exactamente igual a la concentración del anión acetato.

En la siguiente tabla se presentan el nombre de los 20 aminoácidos, con sus abreviaturas de tres y una letra:




 Unión de aminoácidos por enlaces peptídicos en las proteínas:
Unión de aminoácidos por enlaces peptídicos en las proteínas:
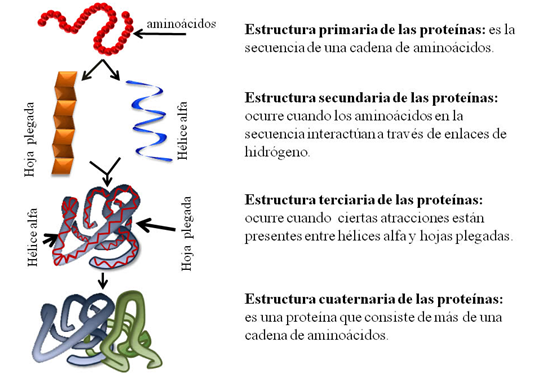
La secuencia lineal de aminoácidos en una cadena polipeptídica se llama estructura primaria de una proteína. A los niveles más altos de estructura se les llaman estructura secundaria: arreglo espacial local de los átomos de la cadena del polipéptido (sin tomar en cuenta la cadena lateral), terciaria: estructura tridimensional de todo el polipéptido y cuaternaria: arreglo espacial de las subunidades de proteínas compuestas por múltiples polipéptidos.

 Las proteínas pueden separarse y caracterizarse por: electroforesis es el desplazamiento de la proteína cargada en un campo eléctrico (gel de poliacrilamida Con SDS, dodecil sulfato sódico).
Las proteínas pueden separarse y caracterizarse por: electroforesis es el desplazamiento de la proteína cargada en un campo eléctrico (gel de poliacrilamida Con SDS, dodecil sulfato sódico).

 Cuando las hebras b son antiparalelas, los puentes de hidrógeno son casi perpendiculares a las cadenas extendidas del polipéptido. Nótese que en la lámina b antiparalela los átomos de oxígeno carbonílico y los de hidrógeno de amida de un residuo forman puentes de hidrógeno con el hidrógeno de amida y el oxígeno carbonílico de un
Cuando las hebras b son antiparalelas, los puentes de hidrógeno son casi perpendiculares a las cadenas extendidas del polipéptido. Nótese que en la lámina b antiparalela los átomos de oxígeno carbonílico y los de hidrógeno de amida de un residuo forman puentes de hidrógeno con el hidrógeno de amida y el oxígeno carbonílico de un
solo residuo en la otra hebra.

Agua
Importancia del agua:
El agua es el líquido más abundante de la corteza y uno de los pocos líquidos naturales. Es esencial en los seres vivos. El agua es el componente más abundante en los medios orgánicos, los seres vivos contienen por término medio un setenta por ciento de agua.
El agua en los seres vivos se encuentra tanto intra como extracelularmente. El agua intracelular es la que está en el interior de las células representa 2/3. El agua extracelular es la que está bañando las células o circulando en forma de sangre, linfa, savia, etc.
Es el componente químico predominante de los organismos vivos.
Presenta singulares propiedades físicas: disuelve una gran cantidad de moléculas orgánicas e inorgánicas, capacidad para formar enlaces de hidrógeno, excelente nucléofilo, es un reactivo o un producto en muchas reacciones metabólicas.
Estructura molecular del agua:

La molécula de agua (H2O) tiene forma de V y el ángulo entre los dos enlaces covalentes O—H es de 104.5°. Algunas propiedades importantes del agua se deben a la forma angulada y a los enlaces intermoleculares que puede formar. El ángulo del enlace H—O-H en el agua es de 104.5°, pero si los orbitales electrónicos apuntaran en realidad a las cuatro esquinas de un tetraedro el ángulo sería de 109.5°. La explicación normal de esta diferencia es que existe una fuerte repulsión entre pares de electrones solitarios y esa repulsión trata de unir los enlaces covalentes, con reducción del ángulo de 109.5° a 104.5°.
El agua es un solvente excelente:
A. Sustancias iónicas y polares se disuelven en agua: El agua puede interactuar y disolver otros compuestos polares y compuestos que se ionizan. La ionización se relaciona con la ganancia o pérdida de un electrón. Las moléculas que se pueden
disociar y formar iones se llaman electrólitos. Las sustancias que se disuelven con facilidad en agua se llaman hidrofílicas o amantes del agua.
disociar y formar iones se llaman electrólitos. Las sustancias que se disuelven con facilidad en agua se llaman hidrofílicas o amantes del agua.
B. Concentraciones celulares y difusión: El comportamiento de los solutos en el citoplasma es distinto del que tienen en una sencilla solución en agua. Una de las diferencias más importantes es la reducción de la velocidad de difusión dentro de las células.
Hay tres razones por las que los solutos se disuelven con más lentitud en las células.
1. La viscosidad del citoplasma es mayor que la del agua, lo que se debe a la
presencia de numerosos solutos, como los azúcares. De acuerdo con mediciones recientes parece que la viscosidad del citoplasma sólo es un poco mayor que la del agua, aun en los organelos empacados densamente.
2. Las moléculas con carga se enlazan momentáneamente entre sí dentro de las
células y ello restringe su movilidad. Dichas consecuencias de la unión ejercen un efecto pequeño, pero apreciable, sobre las tasas de difusión.
3. Los choques con moléculas de agua inhiben la difusión a causa de un efecto
que se denomina hacinamiento molecular. Es la principal razón por la que se
desacelera la difusión en el citoplasma.
1. La viscosidad del citoplasma es mayor que la del agua, lo que se debe a la
presencia de numerosos solutos, como los azúcares. De acuerdo con mediciones recientes parece que la viscosidad del citoplasma sólo es un poco mayor que la del agua, aun en los organelos empacados densamente.
2. Las moléculas con carga se enlazan momentáneamente entre sí dentro de las
células y ello restringe su movilidad. Dichas consecuencias de la unión ejercen un efecto pequeño, pero apreciable, sobre las tasas de difusión.
3. Los choques con moléculas de agua inhiben la difusión a causa de un efecto
que se denomina hacinamiento molecular. Es la principal razón por la que se
desacelera la difusión en el citoplasma.
C. Presión osmótica: Si una membrana permeable al solvente separa a dos soluciones que contienen concentraciones distintas de sustancias disueltas, o solutos, las moléculas del solvente se difundirán desde la solución menos concentrada hacia la más concentrada en un proceso llamado ósmosis. La presión necesaria para evitar este flujo de solvente se llama presión osmótica. La presión osmótica de una solución depende de la concentración molar total del soluto y no de su naturaleza química.
Ionización del agua: Una de las propiedades importantes del agua es su pequeña tendencia a ionizarse. El agua pura no está formada sólo por H2O, sino también por una baja concentración de iones de hidronio y una concentración igual de iones de hidróxido. Los iones hidronio e hidróxido se forman por un ataque nucleofílico del oxígeno contra uno de los protones en una molécula adyacente de agua. La reacción de ionización es una reacción reversible típica. Las reacciones de protonación y desprotonación son muy rápidas. Los iones hidróxido tienen corta duración en el agua, al igual que los iones hidronio. Los iones hidróxido pueden aceptar un protón y convertirse de nuevo en moléculas de agua. A los aceptores de protones se les llama bases. La ionización del agua se puede analizar cuantitativamente. Recuérdese que las
concentraciones de reactivos y productos en una reacción deben llegar al equilibrio. La
relación de esas concentraciones define a la constante de equilibrio (Keq). En el caso de la ionización del agua.
concentraciones de reactivos y productos en una reacción deben llegar al equilibrio. La
relación de esas concentraciones define a la constante de equilibrio (Keq). En el caso de la ionización del agua.
Escala de pH: Existen varios procesos bioquímicos como el transporte de oxígeno en la sangre, la catálisis de reacciones con enzimas y la generación de energía metabólica durante la respiración o la fotosíntesis que están muy influidos por la concentración de protones. Aunque la concentración de H (o H3O) en las células es pequeña en relación con la concentración del agua, el intervalo de [H] en soluciones acuosas es enorme, por lo que conviene usar una cantidad logarítmica llamada pH como medida de la concentración de H. El pH se define como el logaritmo negativo de la concentración de H:
Constantes de disociación de ácidos débiles:
Los ácidos y bases que se disocian por completo en agua, como el ácido clorhídrico y el hidróxido de sodio, se llaman ácidos fuertes y bases fuertes. Hay muchos otros ácidos y bases, como por ejemplo los aminoácidos que forman las proteínas y las purinas y pirimidinas del ADN y ARN, que no se disocian por completo en el agua. La constante de equilibrio para la disociación de un protón de un ácido en agua se
llama constante de disociación del ácido, Ka. Cuando la reacción llega al equilibrio, lo
que sucede con mucha rapidez, la constante de disociación del ácido es igual a la concentración de los productos dividida entre la concentración de los reactivos.
llama constante de disociación del ácido, Ka. Cuando la reacción llega al equilibrio, lo
que sucede con mucha rapidez, la constante de disociación del ácido es igual a la concentración de los productos dividida entre la concentración de los reactivos.
Ecuación de Henderson-Hasselbalch:
Define al pH de una solución en función del pKa del ácido débil en el par ácido-base, y del logaritmo de la relación de las concentraciones de la especie disociada (base conjugada) entre la especie protonada (ácido débil). Nótese que mientras mayor sea la concentración del aceptador de protón (base conjugada) en relación con la del donador de protón (ácido débil) el pH será mayor.
La ecuación de Henderson-Hasselbalch se usa para calcular el pH final de una solución de ácido débil, una vez que la reacción de disociación llega al equilibrio.
Curva de titulación:
Los valores de pKa de los ácidos débiles se determinan por titulación.Ejemplo se titula una solución de ácido acético agregando pequeñas alícuotas de una base fuerte de concentración conocida. Se mide el pH de la solución y se grafica en función de la cantidad de equivalentes molares de base fuerte agregados durante la titulación. Cuando se ha titulado el ácido con la mitad de un equivalente de base, la concentración del ácido acético no disociado es exactamente igual a la concentración del anión acetato.
Los amortiguadores (también llamados disoluciones amortiguadoras, sistemas tampón o buffers) son aquellas disoluciones cuya concentración de protones apenas varía al añadir ácidos o bases fuertes.
Región tampón: es la región de la curva donde existe una mezcla en concentraciones relativamente elevadas del ácido débil y su base conjugada.
Aminoácidos
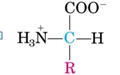
Los aminoácidos son compuestos orgánicos que se combinan para formar proteínas. Los aminoácidos y las proteínas son los pilares fundamentales de la vida. Es una molécula orgánica con un grupo amino (-NH2) y un grupo carboxilo (-COOH). Se pueden dividir en 20 aminoácidos.

En la siguiente tabla se presentan el nombre de los 20 aminoácidos, con sus abreviaturas de tres y una letra:
Estructura de los 20 aminoácidos:
Clasificación de los aminoácidos:
Los aminoácidos se clasifican en: Alifáticos, Polares sin carga (neutros), Átomos de Azufre, Aromáticos, Básicos (carga positiva), Básicos (carga negativa).
Los aminoácidos según su clasificación se pueden agrupar de la siguiente manera:
Ionización de los aminoácidos:
Las propiedades físicas de los aminoácidos reciben influencias de los estados iónicos de los grupos a-carboxilo y a-amino y de todos los grupos ionizables que haya en las cadenas laterales. Cada grupo ionizable guarda relación con un valor específico de pKa, que corresponde al pH al que son iguales las concentraciones de las formas protonada y no protonada. Cuando el pH de la solución es menor que el pKa, predomina la forma protonada y el aminoácido es entonces un ácido real, capaz de donar un protón. Cuando el pH de la solución es mayor que el pKa del grupo ionizable, la forma no protonada de ese grupo predomina, y el aminoácido existe en forma de base conjugada, que es aceptora de protones. Cada aminoácido tiene al menos dos valores de pKa que corresponden a la ionización de los grupos a-carboxilo y a-amino. Los estados iónicos de las cadenas laterales de los aminoácidos influyen sobre las estructuras tridimensionales de las proteínas.
Valores del pKa
| |||
Aminoácido
|
Grupo amino
|
Grupo ácido
|
Cadena lateral
|
Alanina
|
9.9
|
2.4
| |
Valina
|
9.7
|
2.3
| |
Leucina
|
9.7
|
2.3
| |
Isoleucina
|
9.8
|
2.3
| |
Prolina
|
10.6
|
2.0
| |
Triptófano
|
9.4
|
2.5
| |
Fenilalanina
|
9.3
|
2.2
| |
Metionina
|
9.3
|
2.1
| |
Glicina
|
9.8
|
2.4
| |
Serina
|
9.2
|
2.2
| |
Treonina
|
9.1
|
2.1
| |
Cisteína
|
10.7
|
1.9
|
8.4
|
Tirosina
|
9.2
|
2.2
|
10.5
|
Asparagina
|
8.7
|
2.1
| |
Glutamina
|
9.1
|
2.2
| |
Acido Aspártico
|
9.9
|
2.0
|
3.9
|
Acido Glutámico
|
9.5
|
2.1
|
4.1
|
Lisina
|
9.1
|
2.2
|
10.5
|
Histidina
|
9.3
|
1.8
|
12.5
|
Arginina
|
9.0
|
1.8
|
6.0
|
Los valores de pKa de los aminoácidos se determinan con curvas de titulación. El pKa de un grupo ionizable corresponde a un punto medio en su curva de titulación. Es el pH al cual la concentración de la forma ácida (donador de protones) es exactamente igual a la concentración de su base conjugada (aceptor de protones).
La característica más llamativa de los aminoácidos (AA) es la existencia en una misma molécula de grupos ácidos (capaces de ceder H+) y grupos básicos (capaces de captar H+). Por lo tanto, en medio ácido se comportan como bases, y en medio básico se comportan como ácidos. Las moléculas que presentan esta característica se dice que son anfóteros o anfolitos.
Los grupos ácidos y básicos pueden neutralizarse mutuamente, constituyendo una sal interna formada por un ión híbrido (carga positiva y carga negativa), que se llama zwitterión.
Si consideramos un AA sencillo, éste puede adoptar tres formas iónicas diferentes:
El primer grupo que se disocia es el carboxilo (pK1 = 2,22). Por la proximidad del grupo NH3+, el COOH se comporta como un ácido moderadamente fuerte. Si aplicamos la ecuación de Henderson-Hasselbalch al primer equilibrio de disociación, resulta que a pH fisiológico (7,4), la concentración de la forma catiónica es prácticamente despreciable (una de cada 151.000 moléculas en la forma zwitterión). El segundo grupo en disociarse es el NH3+. Como el pK2 es 9,86, la concentración de la forma aniónica es muy pequeña en comparación con la forma zwitterión (1 molécula en forma aniónica por cada 300 en forma zwitteriónica). El AA se comporta a pH fisiológico como un ácido débil que está disociado al 0,35%.
Existe un pH para el cual la carga eléctrica media de las moléculas es cero. Este pH se llama punto isoeléctrico (pI). El pI es el pH en el que la molécula se disocia por igual en ambos sentidos, y como equidista de los dos valores de pK, puede obtenerse por su semisuma:
 Unión de aminoácidos por enlaces peptídicos en las proteínas:
Unión de aminoácidos por enlaces peptídicos en las proteínas: La secuencia lineal de aminoácidos en una cadena polipeptídica se llama estructura primaria de una proteína. A los niveles más altos de estructura se les llaman estructura secundaria: arreglo espacial local de los átomos de la cadena del polipéptido (sin tomar en cuenta la cadena lateral), terciaria: estructura tridimensional de todo el polipéptido y cuaternaria: arreglo espacial de las subunidades de proteínas compuestas por múltiples polipéptidos.
El enlace que se forma entre los aminoácidos es un enlace de amida y se llama enlace peptídico, o enlace de péptido. Esta unión se puede concebir como el resultado de una condensación simple del grupo carboxilo a de un aminoácido con el grupo amino a del otro. A diferencia de los grupos carboxilo y amino de los aminoácidos libres en solución, los grupos que intervienen en los enlaces peptídicos no tienen cargas iónicas.
Las mitades de aminoácido unidas en una cadena polipeptídica se llaman residuos de aminoácido. Los nombres de los residuos se forman sustituyendo la terminación –ina o -ato por -ilo (o -il, en nombres compuestos). La terminación ilo indica que el residuo es una unidad de acilo (estructura que carece del hidroxilo del grupo carboxilo).
Las mitades de aminoácido unidas en una cadena polipeptídica se llaman residuos de aminoácido. Los nombres de los residuos se forman sustituyendo la terminación –ina o -ato por -ilo (o -il, en nombres compuestos). La terminación ilo indica que el residuo es una unidad de acilo (estructura que carece del hidroxilo del grupo carboxilo).
Técnicas de purificación de las proteínas:
Es una serie de procesos que permiten aislar un sólo tipo de proteína de una mezcla compleja. Se pueden aplicar pocas técnicas analíticas en forma directa a las mezclas crudas de proteínas celulares porque contienen cientos (o miles) de proteínas diferentes. Los pasos de purificación son distintos para cada proteína. Se determinan probando con varias técnicas distintas hasta que se desarrolla un procedimiento que produce en forma repetida proteína altamente purificada y que conserva su actividad biológica. Los pasos de purificación suelen aprovechar pequeñas diferencias en las solubilidades, cargas netas, tamaños y especificidades de unión de las proteínas. La mayor parte de las técnicas de purificación se lleva a cabo entre 0 y 4°C para minimizar los procesos dependientes de la temperatura, como la degradación y la desnaturalización (desdoblado) de la proteína. El primer paso en la purificación de una proteína es preparar una solución de proteínas. El siguiente paso de la purificación es, con frecuencia, una relativamente separación cruda, o fraccionamiento, procedimiento que aprovecha las distintas solubilidades de las proteínas en soluciones salinas. Ser reemplazados por los solutos de la solución amortiguadora. A continuación se puede usar la cromatografía en columna para fraccionar la mezcla de proteínas que resta después de la precipitación con sulfato de amonio y la diálisis. Una columna cilíndrica se llena con un material insoluble, como fibras de celulosa sustituida o esferillas de material sintético. La mezcla de proteínas se agrega a la columna y se lava haciendo pasar por la matriz de material insoluble un solvente. A medida que el solvente fluye por la columna, el eluido (que es el líquido que sale por el fondo de la columna) se recolecta en muchas fracciones. Las técnicas cromatográficas se clasifican de acuerdo con el tipo de matriz. En la cromatografía de intercambio iónico la matriz tiene cargas positivas (resinas de intercambio de aniones) o cargas negativas (resinas de intercambio de cationes). La cromatografía por filtración en gel separa a las proteínas con base en su tamaño molecular. El gel es una matriz de esferillas porosas. La cromatografía de afinidad es el tipo de cromatografía en columna más selectivo. Se basa en interacciones específicas de unión entre la proteína deseada y alguna otra molécula enlazada en forma covalente a la matriz de la columna.
Técnicas analíticas:
Las proteínas pueden separarse y caracterizarse por: electroforesis es el desplazamiento de la proteína cargada en un campo eléctrico (gel de poliacrilamida Con SDS, dodecil sulfato sódico).
La espectrometría de masas, como indica el nombre, es una técnica que determina la masa de una molécula. El tipo más sencillo de espectrómetro de masas mide el tiempo que tarda una molécula cargada, en fase gaseosa, para trasladarse desde su punto de inyección hasta un detector sensible. Ese tiempo depende de la carga de una molécula y de su masa, y el resultado se maneja como relación de masa/carga.
En la espectrometría de masas por electroaspersión, la solución de proteína se bombea a través de una aguja metálica, a alto voltaje, para crear diminutas gotitas.
Péptido
Son un tipo de moléculas formadas por la unión de varios aminoácidos mediante enlaces peptídicos.
Estructura tridimensional:
Una conformación es un ordenamiento espacial de átomos que depende de la rotación de uno o varios enlaces. La conformación de una molécula, como la de una proteína, puede cambiar sin que los enlaces covalentes se rompan, mientras que las diversas configuraciones de una molécula sólo se pueden cambiar si se rompen y vuelven a unir enlaces covalentes. La función biológica de una proteína depende por completo de su conformación nativa.
Una proteína puede ser una sola cadena polipeptídica o puede estar formada por varias de esas cadenas unidas entre sí por interacciones débiles. El estudio de grandes conjuntos de proteínas, como el de todo el complemento de proteínas producidas por una célula, es parte de un campo emergente llamado proteómica.
Las proteínas tienen diversas formas. Muchas son macromoléculas aproximadamente esféricas, hidrosolubles y compactas cuyas cadenas polipeptídicas están dobladas de manera apretada. Esas proteínas globulares tienen un interior hidrofóbico y una superficie hidrofílica, en forma característica. Poseen penetraciones o fisuras que reconocen en forma específica a otros compuestos y se unen a ellos en forma transitoria.
Una proteína puede ser una sola cadena polipeptídica o puede estar formada por varias de esas cadenas unidas entre sí por interacciones débiles. El estudio de grandes conjuntos de proteínas, como el de todo el complemento de proteínas producidas por una célula, es parte de un campo emergente llamado proteómica.
Las proteínas tienen diversas formas. Muchas son macromoléculas aproximadamente esféricas, hidrosolubles y compactas cuyas cadenas polipeptídicas están dobladas de manera apretada. Esas proteínas globulares tienen un interior hidrofóbico y una superficie hidrofílica, en forma característica. Poseen penetraciones o fisuras que reconocen en forma específica a otros compuestos y se unen a ellos en forma transitoria.
También los polipéptidos pueden ser partes de grandes estructuras subcelulares o extracelulares, como ribosomas, flagelos y cilios, músculos y cromatina. Las proteínas fibrosas son una clase particular de proteínas estructurales que proporcionan soporte mecánico a las células u organismos. En el caso típico, las proteínas fibrosas se ensamblan en grandes cables o hebras. Como ejemplos de proteínas fibrosas están la a-queratina, el componente principal de cabello y uñas, y la colágena, el componente proteínico principal de tendones, piel, huesos y dientes.
Niveles de estructura de las proteínas:
Métodos para determinar la estructura de las proteínas:
Conformación tridimensional de una proteína se determina por cristalografía con rayos X.
Otra técnica para analizar la estructura macromolecular de las proteínas es la espectroscopia de resonancia magnética nuclear (RMN): En general, los espectros de RMN de pequeñas proteínas, como la ribonucleasa A, se pueden deducir con facilidad, pero el espectro de una molécula grande puede ser en extremo complicado.
Por esta razón es muy difícil determina la estructura de las proteínas mayores,
pero la técnica es muy poderosa para las proteínas más.
Por esta razón es muy difícil determina la estructura de las proteínas mayores,
pero la técnica es muy poderosa para las proteínas más.
Conformación del grupo peptídico:
La estructura de las proteínas comienza con la de los enlaces peptídicos, o enlaces de péptido, que unen a los aminoácidos en una cadena polipeptídica. Los dos átomos que intervienen en el enlace peptídico, junto con sus cuatro sustituyentes (el átomo de oxígeno carbonílico, el átomo de hidrógeno de amida y los dos átomos adyacentes de carbono a) constituyen el grupo peptídico. Los análisis cristalográficos de pequeños péptidos con rayos X revelan que el enlace entre el carbono carbonílico y el nitrógeno es más corto que un enlace sencillo típico C—N, pero más largo que los dobles enlaces CN típicos. Además, el enlace entre el carbono carbonílico y el oxígeno es un poco mayor que el doble enlace típico CO. Esas mediciones indican que los enlaces peptídicos tienen ciertas propiedades del enlace doble y se pueden representar mejor como un híbrido de resonancia. En la conformación trans, los dos carbonos a de residuos adyacentes
de aminoácido están en lados opuestos del enlace peptídico y en las esquinas opuestas
del rectángulo que forma el grupo peptídico plano. En la conformación cis, los dos carbonos a están en el mismo lado del enlace peptídico y están más cerca entre sí. Las conformaciones cis y trans se producen durante la síntesis de la proteína, cuando el enlace peptídico se forma uniendo dos aminoácidos a la cadena polipeptídica. Las dos conformaciones no se pueden interconvertir por giro respecto al enlace peptídico, una vez formado. La conformación cis es menos favorable que la conformación trans, que es
extendida debido a impedimentos estéricos entre las cadenas laterales unidas a los dos
átomos de carbono a. En consecuencia, casi todos los grupos peptídicos en las proteí-
nas tienen la conformación trans.
de aminoácido están en lados opuestos del enlace peptídico y en las esquinas opuestas
del rectángulo que forma el grupo peptídico plano. En la conformación cis, los dos carbonos a están en el mismo lado del enlace peptídico y están más cerca entre sí. Las conformaciones cis y trans se producen durante la síntesis de la proteína, cuando el enlace peptídico se forma uniendo dos aminoácidos a la cadena polipeptídica. Las dos conformaciones no se pueden interconvertir por giro respecto al enlace peptídico, una vez formado. La conformación cis es menos favorable que la conformación trans, que es
extendida debido a impedimentos estéricos entre las cadenas laterales unidas a los dos
átomos de carbono a. En consecuencia, casi todos los grupos peptídicos en las proteí-
nas tienen la conformación trans.
La hélice a:
Una hélice a puede ser una rosca izquierda o derecha. Las hélices a que se encuentran en las proteínas casi siempre son derecha. Los puentes de hidrógeno entre los residuos de aminoácido tienen estabilidad especial en el interior hidrofóbico de una proteína, donde las moléculas de agua no entran y en consecuencia no pueden competir en la formación de puentes. En una hélice a, todos los grupos carbonilo apuntan hacia el C-terminal. Ya que cada grupo peptídico es polar y todos los puentes de hidrógeno apuntan en la misma dirección, toda la hélice es un dipolo con un N-terminal positivo y un C-terminal negativo.
Hebras b y láminas b:
La otra estructura secundaria común se llama estructura b, una clase que incluye a hebras b y láminas b. Las hebras B son partes de la cadena polipeptídica que se encuentran casi totalmente extendidas. Cada residuo en una hebra b ocupa de 0.32 a 0.34 nm de la longitud total, en contraste con la espiral compacta de una hélice a, donde cada residuo corresponde a 0.15 nm de la longitud general. Cuando se ordenan varias hebras b lado a lado forman láminas B. Las hebras b en una lámina pueden ser paralelas (con la misma dirección de N a C-terminal) o antiparalelas (con direcciones opuestas de N a C-terminal).
Cuando las hebras b son antiparalelas, los puentes de hidrógeno son casi perpendiculares a las cadenas extendidas del polipéptido. Nótese que en la lámina b antiparalela los átomos de oxígeno carbonílico y los de hidrógeno de amida de un residuo forman puentes de hidrógeno con el hidrógeno de amida y el oxígeno carbonílico de unsolo residuo en la otra hebra.
Las láminas paralelas son menos estables que las antiparalelas, quizá porque los puentes de hidrógeno están distorsionados en el ordenamiento paralelo. A veces, a la lámina b se le llama lámina B plegada ya que los grupos peptídicos planos se encuentran entre sí formando ángulos como en el plisado de un acordeón.
Asas y giros:
Las asas y los giros unen a hélices a y hebras b y permiten que la cadena de polipéptido se doble sobre sí misma para producir la forma tridimensional compacta que se ve en la estructura nativa. Las asas contienen con frecuencia residuos hidrofílicos y se suelen encontrar en las superficies de las proteínas, donde están expuestas al solvente y forman puentes de hidrógeno con el agua. Las asas que sólo contienen pocos (hasta cinco) residuos se llaman giros si causan un cambio abrupto en la dirección de una cadena de polipéptidos. Los tipos más comunes de giros bruscos se llaman giros inversos o giros B porque con frecuencia conectan hebras b antiparalelas diferentes. Hay dos tipos comunes de giro b que se designan tipo I y tipo II. Ambos contienen cuatro residuos de aminoácidos y los estabilizan puentes de hidrógeno entre el oxígeno carbonílico del primer residuo y el hidrógeno de amida del cuarto residuo. Giro tipo I. La estructura se halla estabilizada por un puente de hidrógeno entre el oxígeno carbonílico del primer residuo N-terminal (Phe) y el hidrógeno de amida del cuarto residuo (Gly). Obsérvese el residuo de prolina en la posición n + 1.
Giro tipo II. También está estabilizado por un puente de hidrógeno entre el oxígeno carbonílico del primer residuo N-terminal (Val) y el hidrógeno de amida del cuarto residuo (Asn).
Giro tipo II. También está estabilizado por un puente de hidrógeno entre el oxígeno carbonílico del primer residuo N-terminal (Val) y el hidrógeno de amida del cuarto residuo (Asn).
Estructura terciaria de las proteínas:
Describe la cadena polipetídica totalmente plegada y compactada (estructura tridimensional del polipéptido). Una propiedad importante de la estructura terciaria es que los residuos de aminoácidos alejados en la estructura primaria se acercan entre sí y permiten interacciones entre sus cadenas laterales. Mientras que la estructura secundaria está estabilizada por puentes de hidrógeno entre los hidrógenos de amida y oxígenos carbonílicos de la columna vertebral del polipéptido, la estructura terciaria se halla estabilizada en especial por interacciones no covalentes.
Estructuras supersecundarias: Las estructuras supersecundarias, o motivos, son combinaciones reconocibles de hélices a, hebras b y giros que aparecen en diversas proteínas. A veces los motivos se relacionan con determinada función, aunque los motivos de estructura similar pueden tener funciones distintas en proteínas diferentes.
Dominios
Hay muchas proteínas que están formadas por varias unidades compactas, discretas,
plegadas en forma independiente llamadas dominios. Los dominios pueden consistir en
combinaciones de motivos. El tamaño de un dominio varía desde unos 25 a 30 residuos
de aminoácidos hasta más de 300. Los dominios
están unidos por asas, pero también se unen entre sí mediante interacciones débiles formadas por las cadenas laterales de aminoácidos en la superficie de cada dominio.
Hay muchas proteínas que están formadas por varias unidades compactas, discretas,
plegadas en forma independiente llamadas dominios. Los dominios pueden consistir en
combinaciones de motivos. El tamaño de un dominio varía desde unos 25 a 30 residuos
de aminoácidos hasta más de 300. Los dominios
están unidos por asas, pero también se unen entre sí mediante interacciones débiles formadas por las cadenas laterales de aminoácidos en la superficie de cada dominio.
Estructura y función de los dominios:
Es compleja la relación entre estructura y función de un dominio. Con frecuencia, un solo dominio tiene determinada función, como por ejemplo unirse a pequeñas moléculas o catalizar una sola reacción. En las enzimas multifuncionales, cada actividad catalítica puede estar asociada con uno de varios dominios presentes en una sola cadena polipeptídica. Las formas únicas de las proteínas, con sus penetraciones, interfases entre dominios y otras grietas les permiten efectuar funciones dinámicas al unirse con otras moléculas en forma selectiva y transitoria.
Es compleja la relación entre estructura y función de un dominio. Con frecuencia, un solo dominio tiene determinada función, como por ejemplo unirse a pequeñas moléculas o catalizar una sola reacción. En las enzimas multifuncionales, cada actividad catalítica puede estar asociada con uno de varios dominios presentes en una sola cadena polipeptídica. Las formas únicas de las proteínas, con sus penetraciones, interfases entre dominios y otras grietas les permiten efectuar funciones dinámicas al unirse con otras moléculas en forma selectiva y transitoria.
Estructura cuaternaria:
Se refiere a la organización y el reordenamiento de las subunidades en una proteína con múltiples sub-unidades. Cada subunidad es una cadena polipeptídica aparte. Los cambios en el ambiente o los tratamientos químicos pueden alterar la conformación nativa de una proteína con la pérdida concomitante de su actividad biológica. Esa alteración se llama desnaturalización. La cantidad de energía necesaria para causar la desnaturalización es pequeña, con frecuencia, quizá la equivalente a la que se necesita para alterar tres o cuatro puentes de hidrógeno.
Propiedades De Las Enzimas











 esta ecuación define a Km como la relación de las constantes de velocidad combinadas para la descomposición de ES dividida entre la constante para su formación.
esta ecuación define a Km como la relación de las constantes de velocidad combinadas para la descomposición de ES dividida entre la constante para su formación.




Las enzimas alostéricas son enzimas cuyas propiedades son afectadas por cambios en la estructura. Los cambios estructurales son ocasionados por interacción con moléculas pequeñas. Una curva de y0 en función de [S] para una enzima alostérica con enlazamiento cooperativo del sustrato. Las curvas sigmoides se deben a la transición entre dos estados de la enzima. En ausencia del sustrato, la enzima está en el estado T. La conformación de cada subunidad presenta una forma en la que se une ineficientemente al sustrato y la velocidad de la reacción es baja. A medida que la concentración de sustrato aumenta, las moléculas de enzima comienzan a unirse al sustrato, aunque la afinidad de la enzima en el estado T sea baja. Cuando una subunidad se une al sustrato sufre un cambio de conformación que la convierte al estado R y se efectúa la reacción. Las propiedades cinéticas de la subunidad enzimática en el estado T y en el estado R son bastante distintas; cada conformación podría, por sí misma, exhibir una cinética normal de Michaelis-Menten.
Propiedades De Las Enzimas
Las enzimas son catalizadores biológicos selectivos de una eficiencia extraordinaria. Toda célula viva dispone de cientos de enzimas distintas que catalizan las reacciones esenciales para la vida. Estas enzimas catalizan las reacciones de las rutas metabólicas centrales, necesarias para mantener la vida.
La mayor parte de las reacciones catalizadas por enzimas no procederían a velocidades apreciables bajo condiciones fisiológicas en ausencia de las enzimas. El papel principal de las enzimas es aumentar las velocidades de tales reacciones. En forma típica, las reacciones catalizadas por las enzimas son de 103 a 1020 veces más rápidas que las mismas sin catalizar.
Un catalizador es una sustancia que acelera la llegada a un equilibrio. Un catalizador puede cambiar en forma temporal durante la reacción, pero no cambia en el proceso general, porque se recicla para participar en varias reacciones.
Los catalizadores aceleran las reacciones tanto hacia adelante como hacia atrás al convertir un proceso de uno o dos pasos en varios pasos menores, cada uno con menor necesidad de energía que la reacción no catalizada.
Las enzimas son muy específicas para los reactivos o sustratos sobre los que actúan, y varía el grado de especificidad hacia el sustrato. Muchas enzimas poseen estereoespecificidad ya que sólo actúan sobre un estereoisómero del sustrato. Quizá el aspecto más importante de la especificidad de una enzima es la especificidad de reacción, esto es, la falta de formación de subproductos como desperdicios. La especificidad de las enzimas no sólo ahorra energía a las células sino que también evita la formación de productos metabólicos potencialmente tóxicos.
Las reacciones acopladas son una propiedad común de muchas enzimas; por ejemplo, la hidrólisis del ATP se acopla con frecuencia a reacciones metabólicas menos favorables.
El nombre enzima deriva de una palabra griega que significa “en la levadura”. Indica que dichos catalizadores están presentes en el interior de las células. A finales del siglo XIX se estudió la fermentación de los azúcares por acción de células de levadura. Una generación después, James B. Summer cristalizó, en 1926, la primera enzima (ureasa) y demostró que era una proteína. En la siguiente década se purificaron cinco enzimas más y se encontró que también eran proteínas: pepsina, tripsina, quimotripsina,
carboxipeptidasa y la enzima Old Yellow (una flavoproteína NADPH oxidasa). Desde entonces se ha demostrado que casi todas las enzimas son proteínas, o proteínas más cofactores. Algunas moléculas de ARN también presentan actividad catalítica, pero usualmente no se les llama enzimas.
Propiedades principales de las enzimas: 1) pueden desempeñarse como catalizadores, 2) catalizan reacciones muy específicas, 3) pueden acoplar reacciones y 4) su actividad puede ser regulada.
Las seis clases de enzimas
Los nombres en la mayor parte de las enzimas metabólicas se forman agregando el sufijo —asa al nombre de sus sustratos, o a un término descriptivo de la reacción que catalizan. Por ejemplo, la ureasa tiene a la urea como sustrato. La alcohol deshidrogenasa cataliza la remoción de hidrógeno de los alcoholes (es decir, la oxidación de alcoholes). Unas pocas enzimas, como la tripsina y la amilasa, se conocen por sus nombres históricos. Las seis categorías son: oxidorreductasas, transferasas, hidrolasas, liasas, isomerasas y ligasas. a cada enxima se le asigna un número único, llamado número de clasificación de la enzima, o número EC (de enzyme classification).
Los nombres en la mayor parte de las enzimas metabólicas se forman agregando el sufijo —asa al nombre de sus sustratos, o a un término descriptivo de la reacción que catalizan. Por ejemplo, la ureasa tiene a la urea como sustrato. La alcohol deshidrogenasa cataliza la remoción de hidrógeno de los alcoholes (es decir, la oxidación de alcoholes). Unas pocas enzimas, como la tripsina y la amilasa, se conocen por sus nombres históricos. Las seis categorías son: oxidorreductasas, transferasas, hidrolasas, liasas, isomerasas y ligasas. a cada enxima se le asigna un número único, llamado número de clasificación de la enzima, o número EC (de enzyme classification).
1. Las oxidorreductasas: catalizan las reacciones de oxidación-reducción. La mayor parte de esas enzimas se llaman, en general, deshidrogenasas. También hay otras enzimas en esta clase que se llaman oxidasas, peroxidasas, oxigenasas o reductasas. Un ejemplo de una oxidorreductasa es la lactato deshidrogenasa (EC 1.1.1.27), llamada también lactato:NAD oxidorreductasa.
2. Las transferasas: catalizan las reacciones de transferencia de un grupo y pueden necesitar la presencia de coenzimas. Este grupo incluye las cinasas, enzimas que catalizan la transferencia de un grupo fosforilo del ATP. La alanina transaminasa, cuyo nombre sistemático es L-alanina:2-oxiglutarato aminotransferasa (EC 2.6.1.2), es un ejemplo típico de esta clase.


3. Las hidrolasas catalizan hidrólisis. Son una clase especial de transferasas donde el agua sirve como aceptor del grupo transferido. La pirofosfatasa es un ejemplo sencillo de una hidrolasa. El nombre sistemático de esta enzima es difosfato fosfohidrolasa (EC 3.6.1.1)
4. Las liasas catalizan la lisis de un sustrato, al generar un enlace doble; son reacciones de eliminación, no hidrolíticas y no oxidantes. Una liasa que cataliza una reacción de adición en las células es frecuentemente llamada sintasa. La piruvato descarboxilasa pertenece a esta clase de enzimas ya que descompone al piruvato en acetaldehído y dióxido de carbono. El nombre sistemático de la piruvato descarboxilasa, 2-oxo-ácido carboxi-liasa (EC 4.1.1.1.) casi nunca se emplea.
5. Las Isomerasas catalizan cambios estructurales dentro de una misma molécula (reacciones de isomerización). La alanina racemasa
(EC 5.1.1.1) es una isomerasa que cataliza la interconversión de L-alanina y D-alanina. El nombre común es igual al nombre sistemático.
6. Las ligasas catalizan la ligadura o unión de dos sustratos. Las ligasas son usualmente llamadas sintetasas. La glutamina
sintetasa, o L-glutamato:amoniaco ligasa (formadora de ADP) (EC 6.3.12) usa la energía de la hidrólisis del ATP para unir glutamato y amoniaco para producir glutamina.

Experimentos Cinéticos:
Cinética Química:
En los experimentos cinéticos se examina la relación entre la cantidad de producto (P) que se forma en una unidad de tiempo ( [P]/ t) y las condiciones experimentales bajo las que se efectúa la reacción. La base de la mayor parte de las mediciones cinéticas es la observación de la rapidez, o velocidad (v), de una reacción, la cual varía en forma directa con la concentración de cada reactante (sección 1.4). Esta observación se expresa en una ecuación de velocidad.
La ecuación de velocidad refleja que la velocidad de una reacción depende de la concentración del sustrato ([S]). El símbolo k es la constante de velocidad e indica la velocidad o la eficiencia de una reacción. Cada reacción tiene una constante de velocidad diferente. La cantidad de producto ([P]) aumenta y la cantidad de sustrato ([S]) disminuye.
Cinética Enzimática:
Emil Fischer, en 1894, propuso que una enzima presenta una plantilla rígida, o cerradura, y que el sustrato es la llave que le corresponde. Los primeros estudios de cinética enzimática confirmaron que una enzima (E) se une a un sustrato para formar un complejo enzima-sustrato (ES). Los complejos ES se forman cuando los ligandos se unen de manera no covalente a sus lugares adecuados en el sitio activo. El sustrato reacciona en forma transitoria con la proteína catalizadora (y con otros sustratos, en una reacción multisustratos) para formar el producto de la reacción.
Esta reacción se efectúa en dos pasos distintos: la formación del complejo enzima-sustrato y la reacción química actual, acompañada por la disociación del producto. La velocidad total de una reacción enzimática depende de las concentraciones tanto del sustrato como del catalizador (la enzima). Cuando la cantidad de enzima es mucho menor que la cantidad de sustrato, la reacción depende de la cantidad de enzima. La concentración de una enzima en una muestra se puede determinar con facilidad comparando su actividad con una curva de referencia. Bajo estas condiciones experimentales hay cantidades suficientes de moléculas de sustrato para que cada molécula de enzima se una a una molécula de sustrato y forme un complejo ES; a esta condición se le llama saturación de la E con el S.
La cinética enzimática se diferencia de la cinética química simple porque las velocidades de reacciones catalizadas por enzimas dependen de la concentración de la enzima y ésta nunca es un producto ni un sustrato de la reacción. Las velocidades también difieren porque el sustrato debe unirse a la enzima para poder convertirse en el producto.
En una reacción catalizada por enzima, las velocidades iniciales se obtienen del progreso de las curvas, igual que en las reacciones químicas.
Ecuación de Michaelis-Menten
Las reacciones catalizadas por enzimas, como cualquier reacción química, se pueden describir en forma matemática como ecuaciones de velocidad. En ellas, varias constantes indican la eficiencia y especificidad de una enzima y en consecuencia son útiles para comparar las actividades de varias enzimas o para evaluar la importancia fisiológica de una determinada enzima.
La pendiente de la curva de y 0 en función de [S] es la de una hipérbola rectangular. Las curvas hiperbólicas son indicativas de procesos donde hay una disociación simple, como se pudo apreciar en la disociación del oxígeno desde la oximioglobina.
La ecuación de una hipérbola rectangular es:
donde a es la asíntota de la curva (el valor de y a un valor de x infinito) y b es el punto del eje x que corresponde a un valor igual a a/2. En los experimentos de cinética enzimática, y y 0 y x [S]. El valor asintótico (a) se llama Vmáx. Es la velocidad máxima de la reacción a concentraciones de sustrato infinitamente grandes.
Una de las características de las curvas hiperbólicas es que parecen aplanarse a concentraciones moderadas de sustrato, a un valor que parece ser mucho menor que la Vmáx. La Vmáx real no se determina tratando de estimar la posición de la asíntota a partir de la forma de la curva. Más bien se determina en forma precisa y correcta ajustando los datos a la ecuación general de una hipérbola rectangular.
El término b en la ecuación general de una hipérbola rectangular se llama constante de Michaelis (Km), y se define como la concentración de sustrato cuando y 0 es igual a la mitad de la Vmáx. La ecuación completa de velocidad es:
El término b en la ecuación general de una hipérbola rectangular se llama constante de Michaelis (Km), y se define como la concentración de sustrato cuando y 0 es igual a la mitad de la Vmáx. La ecuación completa de velocidad es:
La ecuación de Michaelis-Menten describe la relación entre la velocidad inicial de una reacción y la concentración del sustrato.
Deducción dela ecuación de Michaelis-Menten:
Una deducción común de la ecuación de Michaelis-Menten, debida a George E. Briggs y J. B. S. Haldane, es llamada la derivación del estado estable. Esta deducción postula que hay un intervalo de tiempo (llamado estado estable, o estado estacionario) durante el cual se forma el complejo ES a la misma velocidad con la que se descompone, de modo que la concentración de ES es constante. La velocidad inicial se usa en la derivación del estado estable porque se asume que la concentración de producto ([P]) es insignificante.
Al suponer que la concentración de ES en estado estable es constante, entonces la velocidad de formación de producto depende de la velocidad de la reacción química y de la velocidad de disociación de P para abandonar la enzima.
La derivación del estado estable resuelve la ecuación para [ES], usando términos que se pueden medir, como la constante de velocidad, la concentración total de la enzima ([E]total) y la concentración de sustrato ([S]). Se supone que [S] es mayor que [E]total, pero no necesariamente es saturada.
Constante Catalítica
Cuando la concentración de sustrato es alta, la velocidad total de la reacción es Vmáx y está determinada por la concentración de la enzima. La constante de velocidad observada bajo estas condiciones se llama constante catalítica, Kcat, y se define como sigue:
donde Kcat representa la cantidad de moles de sustrato convertidos en producto, por segundo y por mol de enzima (o por mol de sitio activo, para una enzima con multisubunidades) bajo condiciones de saturación. kcat indica la cantidad máxima de moléculas de sustrato convertidas en producto cada segundo por cada sitio activo. A eso se le llama con frecuencia número de recambio. La constante catalítica mide la rapidez con que determinada enzima puede catalizar una reacción específica; es una forma muy útil para describir la eficacia de una enzima. La unidad de kcat es s -1. El recíproco de kcat es el tiempo necesario para que haya un evento catalítico.
Significados de Km
Km también es uno de los parámetros que determina la forma de la curva de y V0 en función de [S]. Es la concentración del sustrato cuando la velocidad inicial es la mitad del valor de Vmáx. Este significado es consecuencia directa de la ecuación general de una hipérbola rectangular.
Las constantes cinéticas indican la actividad enzimática y la eficiencia catalítica
las constantes cinéticas km y kcat se pueden usar para medir las actividades relativas de las enzimas y los sustratos. En la mayor parte de los casos, Km es una medida de la estabilidad del complejo ES, y kcat es similar a la constante de velocidad para la conversión de ES en E P, y cuando el sustrato no es limitante. la velocidad de reacción depende de las concentraciones del sustrato y de la enzima. En términos químicos, es una reacción de segundo orden, y la velocidad depende de una constante de velocidad de segundo orden, que se define por:

Medición de Km y Vmáx
Los parámetros cinéticos de una reacción enzimática pueden producir información valiosa acerca de la especificidad y el mecanismo de la reacción. Los parámetros clave son Km y Vmáx ya que kcat se puede calcular si se conoce Vmáx. Los datos de Km y Vmáx para una reacción catalizada por enzima se pueden determinar de diversas maneras. Para obtener valores fiables de las constantes cinéticas, los puntos de [S]
se deben extender por abajo y por arriba de Km para producir una hipérbola. La ecuación de Michaelis-Menten se puede reacomodar para obtener valores de Vmáx y Km a partir de líneas rectas en gráficas. La transformación de uso más frecuente es la gráfica de doble recíproco, o de Lineweaver-Burk, en la que se grafican los valores de 1/v 0 contra los de 1/[S].
Las constantes cinéticas indican la actividad enzimática y la eficiencia catalítica
las constantes cinéticas km y kcat se pueden usar para medir las actividades relativas de las enzimas y los sustratos. En la mayor parte de los casos, Km es una medida de la estabilidad del complejo ES, y kcat es similar a la constante de velocidad para la conversión de ES en E P, y cuando el sustrato no es limitante. la velocidad de reacción depende de las concentraciones del sustrato y de la enzima. En términos químicos, es una reacción de segundo orden, y la velocidad depende de una constante de velocidad de segundo orden, que se define por:
La relación kcat/Km es útil para comparar las actividades de enzimas diferentes. También es posible evaluar la eficiencia de una enzima midiendo su capacidad catalítica. Este valor es igual a la constante de velocidad de una reacción en presencia de la enzima (kcat/Km) dividida entre la constante de velocidad de la misma reacción en ausencia de la enzima (kn).
Medición de Km y Vmáx
Los parámetros cinéticos de una reacción enzimática pueden producir información valiosa acerca de la especificidad y el mecanismo de la reacción. Los parámetros clave son Km y Vmáx ya que kcat se puede calcular si se conoce Vmáx. Los datos de Km y Vmáx para una reacción catalizada por enzima se pueden determinar de diversas maneras. Para obtener valores fiables de las constantes cinéticas, los puntos de [S]
se deben extender por abajo y por arriba de Km para producir una hipérbola. La ecuación de Michaelis-Menten se puede reacomodar para obtener valores de Vmáx y Km a partir de líneas rectas en gráficas. La transformación de uso más frecuente es la gráfica de doble recíproco, o de Lineweaver-Burk, en la que se grafican los valores de 1/v 0 contra los de 1/[S].
Se pueden obtener valores de kcat con mediciones de Vmáx sólo cuando se conoce la concentración absoluta de la enzima. Se pueden determinar los valores de Km aun con enzimas que no hayan sido purificadas siempre y cuando sea una sola la enzima en la preparación impura la que pueda catalizar la reacción observada.
Cinética de las reacciones con sustratos múltiples
Las mediciones cinéticas de reacciones con sustratos múltiples (o reacciones de multisustrato) son algo más complicadas que para cinéticas enzimáticas sencillas con un solo sustrato. La cinética enzimática simple que se describe en este capítulo se puede ampliar para diferenciar entre varias posibilidades mecanísticas para reacciones multisustrato, como las reacciones de transferencia de grupo.
Las reacciones de multisustrato pueden efectuarse de acuerdo con varios y distintos esquemas cinéticos llamados mecanismos cinéticos porque se deducen en su totalidad mediante experimentos cinéticos. Los mecanismos cinéticos son comúnmente representados con la notación introducida por W. W. Cleland.
Las reacciones consecutivas (o secuenciales) requieren que todos los sustratos estén presentes para que se libere algún producto. Estas reacciones secuenciales pueden ser ordenadas, con un orden obligatorio de enlazamiento de sustratos y de liberación de productos. También pueden ser aleatorias, sin orden obligatorio de enlazamiento o liberación.

En las reacciones ping-pong se libera un producto antes de que se enlacen todos los sustratos. En una reacción ping-pong de bisustrato, se enlaza el primer sustrato, se altera la enzima por sustitución y se libera el primer producto, después de lo cual se une el segundo sustrato, la enzima alterada regresa a su forma original y se libera el segundo producto. al mecanismo de ping-pong se le llama mecanismo de enzima sustituida. La unión y liberación de ligandos en un mecanismo de ping-pong se suelen indicar con líneas inclinadas. Las dos formas de la enzima se representan por E (no sustituida) y F (sustituida).

Inhibición reversible de enzimas
Un inhibidor de enzima (I) es un compuesto que se enlaza con una enzima e interfiere con su actividad. Los inhibidores pueden actuar evitando la formación del complejo ES o bloqueando la reacción química que lleva a la formación del producto. Por regla general, los inhibidores son moléculas pequeñas que se unen en forma reversible con la enzima que inhiben. Los inhibidores artificiales se usan en experimentos para investigar los mecanismos enzimáticos y para descifrar las rutas metabólicas. Algunas medicinas y muchos venenos son inhibidores de enzimas. inhibidores se unen en forma covalente con las enzimas y causando que la inhibición sea irreversible. La mayor parte de la inhibición de relevancia biológica es reversible. Los inhibidores reversibles se unen a las enzimas con las mismas fuerzas no covalentes que enlazan a sustratos y productos. Los inhibidores reversibles se diferencian de los irreversibles por su fácil eliminación de soluciones de enzima por métodos como diálisis o filtración en gel. El equilibrio entre la enzima libre (E) más el inhibidor (I) y el complejo EI se caracteriza por una constante de disociación. En este caso, a la constante se le llama constante de inhibición, Ki.
tipos de inhibición:
Inhibición competitiva:
Los inhibidores competitivos son los que se encuentran con más frecuencia en bioquímica. En la inhibición competitiva, el inhibidor sólo se puede unir a moléculas de enzima libre que no estén unidas a sustrato alguno. Cuando un inhibidor competitivo se une con una molécula de enzima, una molécula de sustrato no puede unirse a esa molécula de enzima. Al revés, la unión de sustrato y una molécula de enzima evita el enlazamiento de un inhibidor. En otras palabras, S e I compiten por unirse a la molécula de enzima. Más comúnmente, S e I se unen al mismo sitio de la enzima, el sitio activo. Este tipo de inhibición se llama inhibición competitiva clásica. el inhibidor se une a un sitio diferente, lo que altera el sitio de unión del sustrato y evita esta unión. A este tipo de inhibición se le llama inhibición competitiva no clásica.
Inhibición acompetitiva:
Los inhibidores acompetitivos sólo se unen al ES y no a la enzima libre. En la inhibición acompetitiva disminuye la Vmáx (aumenta 1/Vmáx) por conversión de algunas moléculas de E en la forma inactiva ESI. Ya que es el complejo ES el que se enlaza con I y la disminución de Vmáx no se revierte por la adición de más sustrato. Los inhibidores acompetitivos hacen descender la Km.
Inhibición no competitiva:
Los inhibidores no competitivos se pueden unir a la E o al ES y formar complejos inactivos EI o ESI, respectivamente. Esos inhibidores no son análogos del sustrato y no se enlazan en el mismo sitio que el S. El caso clásico de inhibición no competitiva se caracteriza por una disminución aparente de Vmáx (1/Vmáx parece aumentar) sin cambiar de la Km. La mayor parte de las enzimas no se apega a la forma clásica de inhibición no competitiva, donde no cambia Km. En la mayoría de los casos se afectan tanto la Vmáx como la Km, ya que la afinidad del inhibidor hacia la E es distinta que hacia ES. En esos casos se suelen llamar de inhibición mixta.
Usos de la inhibición enzimática:
La inhibición enzimática reversible permite contar con un método poderoso para determinar la actividad enzimática y para alterarla en el tratamiento de enfermedades.
La industria farmacéutica recurre a estudios de inhibición enzimática para diseñar medicamentos de uso clínico. En muchos casos se usa un inhibidor natural de una enzima como punto de partida para diseñar un medicamento. En lugar de usar síntesis aleatorias y determinar inhibidores potenciales, algunos investigadores están recurriendo a un método más eficiente llamado diseño racional de un fármaco.
Los progresos en la síntesis de fármacos o medicamentos se ejemplifican por el diseño de una serie de inhibidores de la enzima purina nucleósido fosforilasa. Esta enzima cataliza una reacción de degradación entre fosfato y el nucleósido de guanosina.
Inhibición enzimática irreversible:
un inhibidor enzimático irreversible forma un enlace covalente estable con una molécula de enzima y elimina así las moléculas del sitio activo en la población enzimática. la inhibición irreversible ocurre por alquilación o acilación de la cadena lateral de un residuo de aminoácido en el sitio activo. Una aplicación importante de los inhibidores irreversibles es la identificación de residuos de aminoácidos en el sitio activo, por sustitución específica de sus cadenas laterales reactivas. Los inhibidores reversibles con estructuras que les permitan unirse en forma específica a un sitio activo son más útiles que los reactivos generales de sustitución. Estos inhibidores se llaman reactivos dirigidos al sitio activo, o marcadores de afinidad. El fosfato de bromohidroxiacetona es un marcador de afinidad para la triosa fosfato isomerasa, que cataliza la interconversión de fosfato de dihidroxiacetona y el 3-fosfato de gliceraldehído. Este inhibidor reacciona con la cadena lateral de un residuo de glutamato de la enzima.
Enzimas alostéricas
Las enzimas alostéricas son enzimas cuyas propiedades son afectadas por cambios en la estructura. Los cambios estructurales son ocasionados por interacción con moléculas pequeñas. Una curva de y0 en función de [S] para una enzima alostérica con enlazamiento cooperativo del sustrato. Las curvas sigmoides se deben a la transición entre dos estados de la enzima. En ausencia del sustrato, la enzima está en el estado T. La conformación de cada subunidad presenta una forma en la que se une ineficientemente al sustrato y la velocidad de la reacción es baja. A medida que la concentración de sustrato aumenta, las moléculas de enzima comienzan a unirse al sustrato, aunque la afinidad de la enzima en el estado T sea baja. Cuando una subunidad se une al sustrato sufre un cambio de conformación que la convierte al estado R y se efectúa la reacción. Las propiedades cinéticas de la subunidad enzimática en el estado T y en el estado R son bastante distintas; cada conformación podría, por sí misma, exhibir una cinética normal de Michaelis-Menten.
Regulación de la actividad enzimática:
La cantidad de una enzima se puede controlar regulando la velocidad de su síntesis o de su degradación. Este modo de control se presenta en todas las especies, pero con frecuencia se tarda de muchos minutos a horas sintetizar nuevas enzimas o degradar las enzimas existentes.
En todos los organismos, el control rápido, a escala de segundos o menos, se puede de lograr mediante modulación reversible de la actividad de enzimas reguladoras. Se definen como aquellas cuya actividad se puede modificar en una forma que afecte la velocidad de una reacción catalizada por la enzima. (En muchos casos, tales enzimas reguladoras controlan un paso clave en una ruta metabólica. Cuando se describa la regulación de una ruta, a veces se designarán como enzimas reguladoras a las enzimas que regulan el flujo de metabolitos en una ruta). Las enzimas reguladoras se vuelven catalizadores más activos cuando aumenta la concentración de sus sustratos o cuando disminuyen las concentraciones de los productos de sus rutas metabólicas. Se vuelven menos activas cuando disminuyen las concentraciones de sus sustratos o cuando se acumulan los productos de sus rutas metabólicas.
En todos los organismos, el control rápido, a escala de segundos o menos, se puede de lograr mediante modulación reversible de la actividad de enzimas reguladoras. Se definen como aquellas cuya actividad se puede modificar en una forma que afecte la velocidad de una reacción catalizada por la enzima. (En muchos casos, tales enzimas reguladoras controlan un paso clave en una ruta metabólica. Cuando se describa la regulación de una ruta, a veces se designarán como enzimas reguladoras a las enzimas que regulan el flujo de metabolitos en una ruta). Las enzimas reguladoras se vuelven catalizadores más activos cuando aumenta la concentración de sus sustratos o cuando disminuyen las concentraciones de los productos de sus rutas metabólicas. Se vuelven menos activas cuando disminuyen las concentraciones de sus sustratos o cuando se acumulan los productos de sus rutas metabólicas.
Las enzimas reguladoras se pueden clasificar por el método de su modulación: modulación alostérica no covalente o modificación covalente. Los fenómenos alostéricos son los responsables del control reversible de numerosas enzimas reguladoras. Estas enzimas disponen de un segundo sitio de unión para el ligando, alejado de sus centros catalíticos. Este segundo sitio se llama sitio regulador o sitio alostérico. Un inhibidor o activador alostérico, que también se llama modulador alostérico o efector alostérico, se une al sitio regulador y causa un cambio de conformación en la enzima reguladora.
Propiedades generales de las enzimas alostéricas
- Las actividades de las enzimas alostéricas cambian debido a inhibidores y activadores metabólicos.
- Los moduladores alostéricos se enlazan en forma no covalente a las enzimas que regulan. Muchos moduladores alteran la Km de la enzima para un sustrato; otros, la Vmáx de la enzima.
- Los moduladores mismos no son alterados químicamente por la enzima. Con pocas excepciones, las enzimas reguladoras son proteínas de subunidades múltiples. (Sin embargo, no todas las enzimas de subunidades múltiples son reguladoras). Las cadenas polipeptídicas individuales de una enzima reguladora pueden ser idénticas o diferentes.
- Toda enzima sujeta a regulación alostérica posee cuando menos un sustrato para el cual la curva de v0 en función del [S] es sigmoidea en lugar de hiperbólica.
La teoría concertada, o teoría inducida por simetría, fue inventada para explicar la unión cooperativa de ligandos idénticos como los sustratos.
La teoría secuencial, o teoría inducida por ligando, es una proposición más general. Se basa en la idea de que un ligando puede inducir un cambio en la estructura terciaria de la subunidad a la que se une. Este complejo de subunidad-ligando puede cambiar las conformaciones de las subunidades vecinas hasta diversos grados. La teoría secuencial puede explicar la cooperatividad negativa, una disminución en la afinidad, cuando las moléculas de ligando se unen a un oligómero.
Coenzimas y Vitaminas
Los Cofactores

Las coenzimas: es una molécula inorgánica u orgánica pequeña necesaria para la actividad de una enzima. Las coenzimas funcionan como reactivos de transferencia de grupos.
Las coenzimas funcionan como reactivos de transferencia de grupos. Son especificas para los grupos que acepta y dona.
Hidrógeno
Electrón
Grupos químicos mayores
Muchas enzimas requieren cationes inorgánicos:
 Vitaminas Liposolubles: son las vitaminas que se disuelven en grasas y aceites. Se almacena en cantidades muy abundantes, y esta propiedad les confiere un potencial de toxicidad grave que excede mucho del grupo hidrosoluble.
Se dividen en: Vitamina A, Vitamina D, Vitamina K, Vitamina E.
Vitaminas Liposolubles: son las vitaminas que se disuelven en grasas y aceites. Se almacena en cantidades muy abundantes, y esta propiedad les confiere un potencial de toxicidad grave que excede mucho del grupo hidrosoluble.
Se dividen en: Vitamina A, Vitamina D, Vitamina K, Vitamina E.


NAD y NADP









La coenzima lipoamida es la forma de ácido lipoico unida a proteína. Aunque a menudo se dice que el ácido lipoico es una vitamina B, parece que los animales pueden sintetizarlo.El ácido lipoico es un ácido carboxílico con ocho carbonos (ácido octanoico) en el que se han sustituido dos átomos de hidrógeno, uno en el C-6 y otro en el C-8, por grupos sulfhidrilo en enlaces disulfuro. El ácido lipoico siempre está unido en forma covalente por un enlace de amida, a través de su grupo carboxilo, al grupo e-amino de un residuo de lisina en una proteína. Esta estructura se encuentra en las dihidrolipoamida aciltransferasas, componentes proteínicos del complejo piruvato deshidrogenasa, y en el complejo a-cetoglutarato deshidrogenasa; ambos complejos son de varias enzimas, y están asociados al ciclo del ácido cítrico.





La ubiquinona, llamada también coenzima Q y en consecuencia se abrevia Q, es una coenzima soluble en lípidos, sintetizada por casi todas las especies. Es una benzoquinona con cuatro sustituyentes, uno de los cuales es una larga cadena hidrofóbica. Esta cadena, formada por 6 a 10 unidades de isoprenoides, permite que la ubiquinona se disuelva en los lípidos de las membranas. La ubiquinona es un agente oxidante más enérgico que NAD o que las coenzimas de flavina. En consecuencia, lo pueden reducir NADH o FADH2. Igual que el FMN y el FAD, la ubiquinona puede aceptar o donar dos electrones, uno o dos al mismo tiempo, porque tiene tres estados de oxidación: Q oxidado, radical libre de semiquinona parcialmente reducida y la totalmente reducida QH2, llamada ubiquinol.

Proteínas coenzimas
Los carbohidratos, también conocidos como hidratos de carbono, glúcidos o azucares son sustancias con infinita variedad de propiedades químicas, físicas y fisiológicas. Los carbohidratos (también llamados sacáridos), con base en su masa, son la clase más abundante de moléculas biológicas en la Tierra. Aunque todos los organismos pueden sintetizar carbohidratos, muchos de ellos se producen en organismos fotosintéticos, como bacterias, algas y plantas.Los carbohidratos tienen varios papeles fundamentales en los organismos vivos.Todas las células vivas contienen carbohidratos. Son la principal fuente de energía y la mas rápida. Aportan al organismo entre un 40 y 80 % de los requerimientos totales de energía.








Oligosacárido (Disacárido):




 Es un glicosaminoglicano muy sulfatado, se utiliza ampliamente cómo anticoagulante inyectable, y tiene la densidad de carga más alta conocida de todas las biomoléculas. También se puede utilizar para formar una superficie interior anticoagulante en diversos dispositivos experimentales y médicos tales como tubos de ensayo y máquinas de diálisis renal.
Es un glicosaminoglicano muy sulfatado, se utiliza ampliamente cómo anticoagulante inyectable, y tiene la densidad de carga más alta conocida de todas las biomoléculas. También se puede utilizar para formar una superficie interior anticoagulante en diversos dispositivos experimentales y médicos tales como tubos de ensayo y máquinas de diálisis renal.  Mecanismo de acción
Mecanismo de acción

 La carencia de eritropoyetina ocasiona anemia y como consecuencia los síntomas asociados a ella como debilidad muscular, disminución de la tolerancia al ejercicio físico y mareos.
La carencia de eritropoyetina ocasiona anemia y como consecuencia los síntomas asociados a ella como debilidad muscular, disminución de la tolerancia al ejercicio físico y mareos.
Lípidos y Membrana

 Los ácidos grasos que no contienen dobles enlaces carbono-carbono se llaman saturados, en tanto que los que tienen al menos un doble enlace carbono-carbono se clasifican como no saturados o insaturados. Los ácidos grasos no saturados que sólo tienen un doble enlace carbono-carbono se llaman monoinsaturados, en tanto que los que tienen dos o más se denominan poliinsaturados. La configuración de los dobles enlaces en los ácidos grasos no saturados es cis, en general.
Los ácidos grasos que no contienen dobles enlaces carbono-carbono se llaman saturados, en tanto que los que tienen al menos un doble enlace carbono-carbono se clasifican como no saturados o insaturados. Los ácidos grasos no saturados que sólo tienen un doble enlace carbono-carbono se llaman monoinsaturados, en tanto que los que tienen dos o más se denominan poliinsaturados. La configuración de los dobles enlaces en los ácidos grasos no saturados es cis, en general.













 Esteroides
Esteroides
Es la tercera clase de lípidos que se encuentran en las membranas de los eucariotas y muy rara vez en bacterias.
Los esteroides junto con las vitaminas lipídicas y los terpenos, se clasifican como isoprenoides, por que su molécula se relaciona con le isopreno.
a) Estructura química
b) Esqueleto de carbono
c) Unidad de isopreno
Los esteroides contiene cuatro anillos fundidos,tres de seis carbonos idénticos como A, B y C y un anillo D de cinco carbonos.
La estructura anular característica se deriva del ESCUALENO

















Colestiramina (Questran)
Es una resina polimérica con capacidad para fijar los ácidos biliares. Inicialmente, se utilizó para tratar el prurito secundario a la colestasis, pero hoy día su principal indicación es el tratamiento de la hipercolesterolemia con hipertrigliceridemia concomitante. También se ha utilizado esta resina para tratar la enterocolitis producida por el Clostridium difficile
Dosis:
Mecanismo de acción
Actúa uniéndose a los ácidos biliares e impidiendo su reabsorción. De esta manera se fomenta la transformación del colesterol hepático en ácidos biliares. Secundariamente, la disminución del colesterol incrementa la actividad de los receptores LDL de los hepatocitos, con lo que se incrementa la eliminación del colesterol LDL plasmático.

Vías de administración (formas de uso)
Oral.
 Dosis
Dosis
Absorción
La absorción, al igual que la mayoría de las estatinas, es limitada, reduciéndose a un máximo del 30%. Sufre efecto de primer paso metabólico. Esto junto con la escasa absorción le da una biodisponibilidad muy baja, en torno al 5%. Diversos estudios muestran que su absorción en ayunas es de 2/3 a la registrada inmediatamente después de las comidas.

 La fluvastatina interfiere con la actividad de la hidroximetilglutaril-coenzima A reductasa (HMG-CoA), una enzima hepática, con lo que se interrumpe la vía sintética de la biosíntesis de colesterol en el hombre. Como consecuencia, los niveles del colesterol hepático son reducidos, estimulándose la captación de las LDLs. La fluvastatina reduce el colesterol total circulante, el colesterol asociado a las LDLs y los triglicéridos. La fluvastatina es más potente que la lovastatina como inhibidor de la HMG-CoA reductasa.
La fluvastatina interfiere con la actividad de la hidroximetilglutaril-coenzima A reductasa (HMG-CoA), una enzima hepática, con lo que se interrumpe la vía sintética de la biosíntesis de colesterol en el hombre. Como consecuencia, los niveles del colesterol hepático son reducidos, estimulándose la captación de las LDLs. La fluvastatina reduce el colesterol total circulante, el colesterol asociado a las LDLs y los triglicéridos. La fluvastatina es más potente que la lovastatina como inhibidor de la HMG-CoA reductasa.
 El 28 de febrero de 1953 los científicos James Watson y Francis Crick determinan la estructura del ADN. Los científicos propusieron el modelo de la doble hélice de ADN para representar la estructura tridimensional del polímero.
El 28 de febrero de 1953 los científicos James Watson y Francis Crick determinan la estructura del ADN. Los científicos propusieron el modelo de la doble hélice de ADN para representar la estructura tridimensional del polímero.
En una serie de cinco artículos en el mismo número de Nature se publicó la evidencia experimental que apoyaba el modelo de Watson y Crick.
De éstos, el artículo de Franklin y Raymond Gosling fue la primera publicación con datos de difracción de rayos X que apoyaba el modelo de Watson y Crick, y en ese mismo número de Nature también aparecía un artículo sobre la estructura del ADN de Maurice Wilkins y sus colaboradores.
Watson, Crick y Wilkins recibieron conjuntamente, en 1962, después de la muerte de Rosalind Franklin, el Premio Nobel en Fisiología o Medicina. Sin embargo, el debate continúa sobre quién debería recibir crédito por el descubrimiento.





 La pirimidina tiene un solo anillo de cuatro átomos de carbono y dos de nitrógeno.
La pirimidina tiene un solo anillo de cuatro átomos de carbono y dos de nitrógeno.
La purina tiene un sistema de anillos fundidos de pirimidina y de imidazol. Los dos tiposde bases son no saturados, con dobles enlaces conjugados. Esta propiedad hace que los anillos sean planos, y también explica su capacidad de absorber la luz ultravioleta.











 El ADN de doble hebra es termodinámicamente más estable que las hebras separadas, lo cual explica por qué predomina la forma de doblehebra in vivo. Sin embargo, a veces se puede alterar la estructura de regiones localizadas de la doble hélice al desenrollarse. Esa alteración sucede durante la replicación, reparación, recombinación y transcripción del ADN. Al desenrollamiento y la separación completos de las hebras individuales complementarias se le llama desnaturalización.La desnaturalización sólo sucede in vitro. El ADN de doble hebra se puede desnaturalizar con calor o con un agente
El ADN de doble hebra es termodinámicamente más estable que las hebras separadas, lo cual explica por qué predomina la forma de doblehebra in vivo. Sin embargo, a veces se puede alterar la estructura de regiones localizadas de la doble hélice al desenrollarse. Esa alteración sucede durante la replicación, reparación, recombinación y transcripción del ADN. Al desenrollamiento y la separación completos de las hebras individuales complementarias se le llama desnaturalización.La desnaturalización sólo sucede in vitro. El ADN de doble hebra se puede desnaturalizar con calor o con un agente
caotrópico, como urea o cloruro de guanidinio. En estudios de desnaturalización térmica se aumenta lentamente la temperatura de una solución de ADN. Al elevar la temperatura, se dispersan cada vez más bases y se rompen más puentes de hidrógeno entre pares de bases. Llega un momento en que las dos cadenas se separan por completo. La temperatura a la que la mitad del ADN se ha convertido en una sola cadena se le llama punto de fusión, Tm.
Para medir el grado de desnaturalización se usa la absorción de luz ultravioleta. Se hacen mediciones a una longitud de onda de 260 nm, cercana al máximo de absorbencia para los ácidos nucleicos. El ADN de una hebra absorbe 12 a 14% más luz que el ADN de doble hebra a 260 nm. Una gráfica del cambio de absorbencia de una solución de ADN en función de la temperatura se llama curva de fusión. La absorbencia aumenta en forma marcada en el punto de fusión, y la transición de ADN de doble hebra a una hebra se efectúa dentro de límites estrechos de temperatura.

 Una molécula circular de ADN con la conformación B tiene un promedio de 10.4 pares de bases por vuelta. Se dice que está relajada si tal molécula puede reposar plana sobre una superficie. Esta doble hélice relajada se puede seguir envolviendo o desenvolviendo si se rompen las hebras del ADN y se tuercen los dos extremos de la molécula lineal en direcciones opuestas. Cuando se vuelven a unir las hebras para crear un círculo, ya no hay 10.4 pares de bases por vuelta, las necesarias para mantener la conformación B estable. La molécula circular se compensa por el envolvimiento o desenvolvimiento formando superenrollamientos que restauran 10.4 pares de bases por vuelta de la doble hélice. Cada superenrollamiento se compensa por una vuelta de la doble hélice.
Una molécula circular de ADN con la conformación B tiene un promedio de 10.4 pares de bases por vuelta. Se dice que está relajada si tal molécula puede reposar plana sobre una superficie. Esta doble hélice relajada se puede seguir envolviendo o desenvolviendo si se rompen las hebras del ADN y se tuercen los dos extremos de la molécula lineal en direcciones opuestas. Cuando se vuelven a unir las hebras para crear un círculo, ya no hay 10.4 pares de bases por vuelta, las necesarias para mantener la conformación B estable. La molécula circular se compensa por el envolvimiento o desenvolvimiento formando superenrollamientos que restauran 10.4 pares de bases por vuelta de la doble hélice. Cada superenrollamiento se compensa por una vuelta de la doble hélice.
Transcripción y procesamiento del ARN

 El fármaco es usado para tratar el HIV tipo 1 y debe siempre ser utilizado en combinación con otros agentes antirretrovirales. Abacavir jamás debe usarse como único fármaco cuando se cambien los regímenes antirretrovirales debido a pérdida de la respuesta viral.
El fármaco es usado para tratar el HIV tipo 1 y debe siempre ser utilizado en combinación con otros agentes antirretrovirales. Abacavir jamás debe usarse como único fármaco cuando se cambien los regímenes antirretrovirales debido a pérdida de la respuesta viral.
 La nevirapina (BI-RG-587) es un inhibidor de la transcriptasa inversa no nucleósido (INNTI) con actividad frente al VIH-1. Estructuralmente es un miembro del grupo de las dipiridodiazepinonas (C15H14N4O; PM 266.3). La nevirapina actúa por un mecanismo de inhibición no competitiva reversible (1). Se une directamente a la trascriptasa inversa (TI) causando una alteración del lugar catalítico del enzima (2) y bloqueando las actividades de la ADN polimerasa, ARN-dependientes y ADN-dependientes.
La nevirapina (BI-RG-587) es un inhibidor de la transcriptasa inversa no nucleósido (INNTI) con actividad frente al VIH-1. Estructuralmente es un miembro del grupo de las dipiridodiazepinonas (C15H14N4O; PM 266.3). La nevirapina actúa por un mecanismo de inhibición no competitiva reversible (1). Se une directamente a la trascriptasa inversa (TI) causando una alteración del lugar catalítico del enzima (2) y bloqueando las actividades de la ADN polimerasa, ARN-dependientes y ADN-dependientes.
 Dosis:
Dosis:

Vías de administración
Mecanismo de Acción
 El aciclo-GTP inhibe la síntesis de ADN viral a través de un mecanismo competitivo con la polimerasa viral, y al ser incorporada en la cadena de ADN en síntesis, detiene su replicación. Su poca afinidad a las polimerasas celulares, sumado al hecho de que la fosforilación ocurre sólo en células infectadas, hace que tenga una toxicidad baja.
El aciclo-GTP inhibe la síntesis de ADN viral a través de un mecanismo competitivo con la polimerasa viral, y al ser incorporada en la cadena de ADN en síntesis, detiene su replicación. Su poca afinidad a las polimerasas celulares, sumado al hecho de que la fosforilación ocurre sólo en células infectadas, hace que tenga una toxicidad baja.
Coenzimas y Vitaminas
Los Cofactores
son componente no proteico, termoestable y de baja masa molecular, necesario para la acción de una enzima. El cofactor se une a una estructura proteica, denominada apoenzima, y el complejo apoenzima-cofactor recibe el nombre de holoenzima.
Hay dos tipos de cofactores: los iones esenciales (principalmente iones metálicos) y los compuestos orgánicos llamados coenzimas. Los cofactores, tanto inorgánicos como orgánicos, se transforman en partes esenciales de los sitios activos de ciertas enzimas.
Las coenzimas funcionan como reactivos de transferencia de grupos. Son especificas para los grupos que acepta y dona.
Hidrógeno
Electrón
Grupos químicos mayores
Muchas enzimas requieren cationes inorgánicos:
Más de la cuarta parte de todas las enzimas conocidas requieren cationes metálicos para tener actividad catalítica total. Estas enzimas se pueden dividir en dos grupos: enzimas activadas por metal y metaloenzimas. Las enzimas activadas por metal tienen necesidad absoluta de iones metálicos adicionales, o son estimuladas por adición de iones metálicos. Algunas de esas enzimas requieren cationes monovalentes, como K y otras necesitan cationes divalentes, como Ca o Mg. Las metaloenzimas contienen iones metálicos firmemente unidos en sus sitios activos. Los iones que más se suelen encontrar en las metaloenzimas son de metales de transición como hierro y zinc, y con menos frecuencia cobre y cobalto. Los iones metálicos que se unen fuertemente a las enzimas con frecuencia tienen funciones predominantes en la catálisis. Los iones de algunas metaloenzimas pueden funcionar como catalizadores electrofílicos.
Clasificación de las coenzimas:
Las coenzimas se clasifican en :
Cosustratos: con frecuencia son sustratos en reacciones catalizadas por enzimas. Un cosustrato se altera durante la reacción y se disocia del sitio activo. La estructura original del cosustrato se regenera en una reacción posterior, catalizada por la enzima. El cosustrato se recicla en la célula.
Los cosustratos llevan y traen grupos metabólicos entre distintas reacciones catalizadas por enzimas.
Grupos prostéticos: permanece unido a la enzima durante la reacción. Generalmente se unen de manera covalente a la enzima o puede estar unido por muchas interacciones débiles.
Vitaminas:
Fue acuñada por Casimir Funk en 1912 para describir una "amina vital" de los hollejos del arroz, que curaba el beriberi, una enfermedad por deficiencia nutricional que causa degeneración neural. El beriberi fue descrito primero en los pájaros, y después en humanos cuyas dietas consistían principalmente en arroz limpio (pulido). La sustancia anti-beriberi (tiamina) se conoció como vitamina B1.
Las Vitaminas se clasifican en:
· Vitaminas Hidrosolubles: son las vitaminas son las que se disuelven en agua. Se almacenan en una cantidad limitada, y se requiere consumo frecuente para conservar la saturación de los tejidos. Se dividen en: Complejo B (B1, B2, B3, B5, B6, B12, etc.) y Acido Ascórbico. el complejo B comprende muchos compuestos que muestran grandes diferencias en su estructura y efecto biológico. El Acido Ascórbico o Vitamina C se encuentra concentrada especialmente en frutas cítricas.


Algunas vitaminas y sus enfermedades relacionadas por deficiencia en la dieta

ATP y otros cosustratos nucleótidos
Hay varios trifosfatos nucleósidos que son coenzimas. Con mucho, el más abundante es el trifosfato de adenosina (ATP, adenosine triphosphate). Entre otros ejemplos frecuentes están el GTP, la S-adenosilmetionina y azúcares nucleótido, como la difosfato de uridina glucosa (UDP-glucosa). El ATP (figura 7.4) es un reactivo versátil que puede donar sus grupos fosforilo, pirofosforilo, adenililo (AMP) o adenosilo en reacciones de transferencia de grupo. La reacción más común donde interviene el ATP es la transferencia del grupo fosforilo.
El ATP también es la fuente de otras coenzimas metabólicas. Una es la S-adenosilmetionina (figura 7.5), y es sintetizada por la reacción de metionina con ATP.
Metionina + ATP → S-Adenosilmetionina Pi PPi
NAD y NADP
Las coenzimas de nicotinamida son nicotinamida adenina dinucleótido (NAD ) y el fosfato de nicotinamida adenina dinucleótido (NADP ), muy relacionado. Fueron las primeras coenzimas que se conocieron. Ambas contienen nicotinamida, la amida del ácido nicotínico. El ácido nicotínico (llamado también niacina) es el factor que falta en la pelagra. Es esencial como precursor de NAD y NADP . (En muchas especies, el triptófano se degrada a ácido nicotínico. En consecuencia, el triptófano en la dieta puede suplir algo de los requisitos de niacina o nicotinamida). Como el ácido nicotínico es el derivado de piridina con 3-carboxilo, con frecuencia se llaman coenzimas nucleótidos de piridina a las coenzimas de nicotinamida. Las coenzimas de nicotinamida participan en muchas reacciones de oxidación-reducción. Ayudan en la transferencia de electrones hacia metabolitos y desde éstos. Las formas oxidadas, NAD y NADP , carecen de electrones, y las formas reducidas, NADH y NADPH, tienen un par adicional de electrones en forma de un ion hidruro unido en forma covalente.
Estructura de las coenzimas:

Formas oxidada y reducida de NAD (y de NADP). El anillo de piridina de la se reduce por la adición de un ion hidruro al C-4 cuando la se convierte en NADH (y cuando el NADP se convierte en NADPH). En el se fosforila el grupo 2 -hidroxilo del anillo de azúcar en la adenosina. El centro reactivo de estas coenzimas se muestra en gris oscuro.
FAD y FMN
Las coenzimas flavina adenina dinucleótido (FAD) y flavina mononucleótido (FMN) se derivan de la riboflavina o vitamina B2. La riboflavina es sintetizada por bacterias, protistas, hongos, plantas y algunos animales. Los mamíferos obtienen riboflavina de su alimento. La riboflavina está formada por ribitol, un alcohol con cinco carbonos unido al átomo N-10 de un sistema de anillo heterocíclico llamado isoaloxazina. Igual que el NAD y el el FAD contiene AMP y un enlace de pirofosfato.
El FAD y FMN se reducen a FADH2 y FMNH2, tomando un protón y dos electrones en forma de un ion hidruro. Las enzimas oxidadas son amarillo brillante, como resultado del sistema de doble enlace conjugado del sistema anular de isoaloxazina. El color se pierde cuando las coenzimas se reducen a FMNH2 y FADH2. A diferencia de la NADH y NADPH, que participan sólo en la transferencia de doselectrones, las FMNH2 y FADH2 donan electrones, uno por uno, o dos al mismo tiempo. El compuesto FADH• o FMNH•, parcialmente oxidado, se forma cuando se dona un electrón. Estos intermediarios son radicales libres relativamente estables, llamados semiquinonas.
Coenzima A
Muchos procesos metabólicos dependen de la coenzima A (CoA o HS-CoA), como laoxidación de moléculas de combustible y la biosíntesis de algunos carbohidratos y lípidos. Esta coenzima interviene en reacciones de transferencia de grupo acilo, donde los grupos metabólicos móviles son ácidos carboxílicos y ácidos grasos simples. La coenzima A tiene dos componentes principales: una unidad de 2 mercaptoetilamina, que tiene un grupo —SH libre, la vitamina pantotenato— (vitamina B5, una amida de β-alanina y pantoato), y una mitad de ADP, cuyo grupo hidroxilo 3 está esterificado con un tercer grupo fosfato. El centro reactivo de la CoA es el grupo —SH. Los grupos acilo se unen en forma covalente al grupo —SH para formar tioésteres. La fosfopanteteína, un éster de fosfato que contiene las mitades de 2-mercaptoetilamina y pantotenato de la coenzima A, es el grupo prostético de una pequeña proteína (de 77 residuos de aminoácidos) llamada proteína portadora de acilo (ACP, acyl carrier protein). El grupo prostético se esterifica formando ACP a través del oxígeno en la cadena lateral de un residuo de serina.

Pirofosfato de tiamina:
La tiamina (o vitamina B1) contiene un anillo de pirimidina y un anillo de tiazolio con carga positiva. En los mamíferos, la tiamina es una vitamina esencial. Abunda en las cáscaras de arroz y en otros cereales. La coenzima es el pirofosfato de tiamina (TPP, thiamine pyrophosphate). El TPP se sintetiza a partir de la tiamina, por transferencia enzimática de un grupo pirofosforilo del ATP.
También el TPP es una coenzima que interviene en la descarboxilación oxidante de α-cetoácidos distintos del piruvato. Además, el TPP es un grupo prostético para las enzimas llamadas transcetolasas, que catalizan la transferencia de grupos de dos carbonos que contienen un anillo ceto, entre moléculas de azúcar.
Fosfato de piridoxal
La familia de vitaminas B6 hidrosolubles consiste en tres moléculas estrechamente relacionadas que sólo difieren en el estado de oxidación o aminación en el carbono unido a la posición 4 del anillo de piridina. La vitamina B6, con mayor frecuencia piridoxal o piridoxamina, se encuentra con facilidad en muchas fuentes vegetales y animales. Las deficiencias de B6 inducidas en ratas causan dermatitis y diversas alteraciones relacionadas con el metabolismo de las proteínas, pero en realidad son raras las deficiencias de B6 en humanos. El fosfato de piridoxal es el grupo prostético de muchas enzimas que catalizan diversas reacciones donde intervienen aminoácidos, como isomerizaciones, descarboxilaciones y eliminaciones o sustituciones de cadena lateral.

Biotina
La biotina es un grupo prostético para enzimas que catalizan reacciones de transferencia del grupo carboxilo y reacciones de carboxilación dependientes de ATP. La biotina está unida en forma covalente al sitio activo de su enzima anfitriona por un enlace de amida al grupo e-amino de un residuo de lisina.
La biotina se identificó por primera vez como factor esencial para el crecimiento de las levaduras. Como la biotina es sintetizada por las bacterias intestinales, y sólo se requiere en muy pequeñas cantidades (microgramos por día), es rara la deficiencia de biotina en los humanos o animales alimentados con dietas normales. Sin embargo, se puede inducir una deficiencia de biotina ingiriendo claras de huevo crudas, que contienen una proteína llamada avidina. La avidina se une fuertemente a la biotina y la hace no disponible para absorción en el tracto intestinal. Cuando se cocinan los huevos, la avidina se desnaturaliza y pierde su afinidad hacia la biotina.
Tetrahidrofolato
La vitamina folato se aisló por primera vez a principios de la década de 1940, a partir de hojas verdes, hígado y levadura. El folato tiene tres componentes principales: pterina (2-amino-4-oxopteridina), una mitad de ácido p-aminobenzoico, y un residuo de glutamato.
Los humanos requieren folato en su dieta, porque no pueden sintetizar el compuesto pterina-ácido p-aminobenzoico (PABA, p-aminobenzoic acid), y no se puede adicionar glutamato a PABA exógeno.
La estructura de la coenzima folato, que se llaman tetrahidrofolato en forma colectiva, difieren de la vitamina en dos cosas: son compuestos reducidos (5,6,7,8-tetrahidropterinas) y están modificadas por adición de residuos de glutamato unidos entre sí mediante enlaces de g-glutamilamida.
Los humanos requieren folato en su dieta, porque no pueden sintetizar el compuesto pterina-ácido p-aminobenzoico (PABA, p-aminobenzoic acid), y no se puede adicionar glutamato a PABA exógeno.
La estructura de la coenzima folato, que se llaman tetrahidrofolato en forma colectiva, difieren de la vitamina en dos cosas: son compuestos reducidos (5,6,7,8-tetrahidropterinas) y están modificadas por adición de residuos de glutamato unidos entre sí mediante enlaces de g-glutamilamida.

La principal función metabólica de la dihidrofolato reductasa es la reducción del hidrofolato producido durante la formación del grupo metilo en el timidilato (dTMP).
Cobalamina
La cobalamina (vitamina B2) es la mayor de las vitaminas B, y fue la última que se aisló.
 |
| Estructura de la cobalamina |
Algunas especies de bacterias sintetizan la cobalamina. Todos los animales la requieren como micronutriente, y también algunas bacterias y algas. Las plantas no requieren cobalamina, por lo que no la sintetizan. En consecuencia, y en el caso normal, los humanos obtienen la vitamina B12 a partir de alimentos de origen animal. La deficiencia de cobalamina puede causar anemia perniciosa, enfermedad potencialmente fatal debida a una disminución en la producción de glóbulos rojos por la médula ósea.
Lipoamida
La coenzima lipoamida es la forma de ácido lipoico unida a proteína. Aunque a menudo se dice que el ácido lipoico es una vitamina B, parece que los animales pueden sintetizarlo.El ácido lipoico es un ácido carboxílico con ocho carbonos (ácido octanoico) en el que se han sustituido dos átomos de hidrógeno, uno en el C-6 y otro en el C-8, por grupos sulfhidrilo en enlaces disulfuro. El ácido lipoico siempre está unido en forma covalente por un enlace de amida, a través de su grupo carboxilo, al grupo e-amino de un residuo de lisina en una proteína. Esta estructura se encuentra en las dihidrolipoamida aciltransferasas, componentes proteínicos del complejo piruvato deshidrogenasa, y en el complejo a-cetoglutarato deshidrogenasa; ambos complejos son de varias enzimas, y están asociados al ciclo del ácido cítrico.

Vitaminas lipídicas
Las estructuras de las cuatro vitaminas lipídicas (A, D, E y K) contienen anillos y largas cadenas laterales alifáticas. Las vitaminas lipídicas son muy hidrofóbicas, aunque cada una posee cuando menos un grupo polar. Al ingerirse son absorbidas en el intestino por un proceso parecido a la absorción de otros nutrientes lípidos.
Las estructuras de las cuatro vitaminas lipídicas (A, D, E y K) contienen anillos y largas cadenas laterales alifáticas. Las vitaminas lipídicas son muy hidrofóbicas, aunque cada una posee cuando menos un grupo polar. Al ingerirse son absorbidas en el intestino por un proceso parecido a la absorción de otros nutrientes lípidos.
Vitamina A
La vitamina A, o retinol, es una molécula lipídica con 20 carbonos, que se obtiene en la dieta, ya sea en forma directa o indirecta, como b-caroteno. Las zanahorias y otras verduras amarillas son ricas en b-caroteno, un lípido vegetal con 40 carbonos cuya ruptura oxidante enzimática produce la vitamina A.
La vitamina A existe en tres formas que difieren en estado de oxidación del grupo funcional terminal: el retinol, un alcohol estable, el retinal, un aldehído, y el ácido retinoico. Los tres compuestos tienen funciones biológicas importantes. El ácido retinoico es un compuesto señalador que se une a proteínas receptoras dentro de las células; los complejos ligando-receptor se unen entonces a cromosomas y pueden regular la expresión genética durante la diferenciación celular. El aldehído retinal es un compuesto sensible a la luz, con importante papel en la visión. El retinal es el grupo prostético de la proteína rodopsina, y la absorción de un fotón de luz en el retinal dispara un impulso nervioso.

Vitamina D
La vitamina D es un nombre colectivo de un grupo de lípidos relacionados. Cuando los humanos se exponen a suficiente luz solar, se forma vitamina D3 (colecalciferol) en forma no enzimática, en la piel, a partir del esteroide 7-dehidrocolesterol. La vitamina D2, compuesto relacionado con la vitamina D3 (la D2 tiene un grupo metilo adicional) es el aditivo en leches fortificadas. La forma activa de la vitamina D3, el 1,25-dihidroxicolecalciferol, se forma a partir de la vitamina D3 mediante dos reacciones de hidroxilación
La vitamina D es un nombre colectivo de un grupo de lípidos relacionados. Cuando los humanos se exponen a suficiente luz solar, se forma vitamina D3 (colecalciferol) en forma no enzimática, en la piel, a partir del esteroide 7-dehidrocolesterol. La vitamina D2, compuesto relacionado con la vitamina D3 (la D2 tiene un grupo metilo adicional) es el aditivo en leches fortificadas. La forma activa de la vitamina D3, el 1,25-dihidroxicolecalciferol, se forma a partir de la vitamina D3 mediante dos reacciones de hidroxilación

Vitamina E
La vitamina E, o a-tocoferol es uno de varios tocoferoles estrechamente relacionados; son compuestos que tienen un sistema anular bicíclico oxigenado, con una cadena lateral hidrofóbica. El grupo fenol de la vitamina E puede oxidarse y formar un radical libre estable. Se cree que la vitamina E funciona como agente reductor que secuestra oxígeno y radicales libres; esta acción oxidante podría evitar daños a los ácidos grasos en las membranas biológicas.
La vitamina E, o a-tocoferol es uno de varios tocoferoles estrechamente relacionados; son compuestos que tienen un sistema anular bicíclico oxigenado, con una cadena lateral hidrofóbica. El grupo fenol de la vitamina E puede oxidarse y formar un radical libre estable. Se cree que la vitamina E funciona como agente reductor que secuestra oxígeno y radicales libres; esta acción oxidante podría evitar daños a los ácidos grasos en las membranas biológicas.

Vitamina KLa vitamina K (filoquinona) es una vitamina lipídica procedente de plantas, necesaria en la síntesis de algunas de las proteínas que intervienen en la coagulación sanguínea. Es una coenzima de una carboxilasa de mamíferos que cataliza la conversión de residuos específicos de glutamato para formar residuos de g-carboxiglutamato. La forma reducida (hidroquinona) de la vitamina K participa en la carboxilación como agente reductor.

Ubiquinona
La ubiquinona, llamada también coenzima Q y en consecuencia se abrevia Q, es una coenzima soluble en lípidos, sintetizada por casi todas las especies. Es una benzoquinona con cuatro sustituyentes, uno de los cuales es una larga cadena hidrofóbica. Esta cadena, formada por 6 a 10 unidades de isoprenoides, permite que la ubiquinona se disuelva en los lípidos de las membranas. La ubiquinona es un agente oxidante más enérgico que NAD o que las coenzimas de flavina. En consecuencia, lo pueden reducir NADH o FADH2. Igual que el FMN y el FAD, la ubiquinona puede aceptar o donar dos electrones, uno o dos al mismo tiempo, porque tiene tres estados de oxidación: Q oxidado, radical libre de semiquinona parcialmente reducida y la totalmente reducida QH2, llamada ubiquinol.
Algunas proteínas funcionan como coenzimas. No catalizan reacciones ellas mismas, pero ciertas enzimas las necesitan. Esas coenzimas se llaman proteínas de transferencia de grupo, o proteínas coenzimas. Contienen un grupo funcional que es parte de la columna vertebral de la proteína, o bien es un grupo prostético. En general son más pequeñas y más termoestables que la mayor parte de las enzimas. Las proteínas coenzimas se llaman coenzimas porque participan en muchas y diversas reacciones, y se asocian a una variedad de enzimas diferentes.
Citocromos
Los citocromos son proteínas coenzimas que contienen hemo, cuyos átomos de Fe(III) sufren reducción reversible de un electrón. Los citocromos pueden ser de clase a, b y c, de acuerdo con sus espectros de absorción visible. Las clases tienen grupos hemo prostéticos un poco diferentes. El grupo hemo de los citocromos tipo b es igual al de hemoglobina y mioglobina. El hemo del citocromo a tiene una cadena hidrofóbica de 17 carbonos en el C-2 del anillo de porfirina, y un grupo formilo en el C-8, en tanto que el hemo tipo b tiene un grupo vinilo unido al C-2 y un grupo metilo en el C-8. En los citocromos tipo c, el hemo está unido en forma covalente a la apoproteína por grupos vinilo del hemo mediante dos enlaces tioéter que se forman por adición de los grupos tiol de dos residuos de cisteína.
Los citocromos son proteínas coenzimas que contienen hemo, cuyos átomos de Fe(III) sufren reducción reversible de un electrón. Los citocromos pueden ser de clase a, b y c, de acuerdo con sus espectros de absorción visible. Las clases tienen grupos hemo prostéticos un poco diferentes. El grupo hemo de los citocromos tipo b es igual al de hemoglobina y mioglobina. El hemo del citocromo a tiene una cadena hidrofóbica de 17 carbonos en el C-2 del anillo de porfirina, y un grupo formilo en el C-8, en tanto que el hemo tipo b tiene un grupo vinilo unido al C-2 y un grupo metilo en el C-8. En los citocromos tipo c, el hemo está unido en forma covalente a la apoproteína por grupos vinilo del hemo mediante dos enlaces tioéter que se forman por adición de los grupos tiol de dos residuos de cisteína.
Resumen de las vitaminas y formas coenzimáticas
|



Composición de los carbohidratos:
- Están formados por carbono (C), hidrogeno (H) y oxigeno (O).
- Presentan la formula química general (CH2O)n.
- Todos los carbohidratos presentan grupos funcionales -C=O o –OH.
- Los carbohidratos se encuentran de forma abundante en las plantas y en pequeñas cantidades en animales.
- Las plantas nos proveen de todos los hidratos de carbono mediante la alimentación.
Aldosas: Los aldehídos este grupo carbonilo se encuentra en un extremo de la cadena hidrocarbonada, por lo que tiene un átomo de hidrógeno unido a él directamente, que sueleescribirse, por comodidad, en la forma —CHO. Según el tipo de grupo hidrocarbonado unido al grupo funcional, los aldehídos pueden ser alifáticos, R—CHO, y aromáticos, Ar— CHO. Gliceraldehido es un aldehído y estos monosacáridos se denominamos aldosas.



Cetosas: Las cetonas el grupo carbonilo se encuentra unido a dos grupos hidrocarbonados. Según el tipo de grupo hidrocarbonado unido al grupo funcional, las cetonas se clasifican en alifáticas, R—CO—R', aromáticas, Ar—CO—Ar, y mixtas; R—CO—Ar. Dihidroxiacetona es una cetona y estos monosacáridos se denominan cetosas.

Estereoisomeros:
Es un isómero que tiene la misma fórmula molecular y cuadricula, también la misma secuencia de átomos enlazados, con los mismos enlaces entre sus átomos, pero difieren en la orientación tridimensional de sus átomos en el espacio.

En general, hay 2n estereoisomeros posibles de un compuesto que tenga n carbonos quirales (un carbono quiral es un átomo de carbono que está enlazado con cuatro sustituyentes o elementos diferentes. Puede presentarse en algunos compuestos orgánicos, es decir, en aquellos que están presentes en los seres vivos, como los glúcidos).
Epímeros: Cuando las moléculas de azúcar tienen distinta configuración sólo en uno de varios centros quirales.
Clasificación de los Carbohidratos según el numero de unidades de carbohidrato:
- Monosacárido
- Oligosacárido (Disacárido)
- Polisacárido
Los monosacáridos: Unidad mas pequeña de estructura de los carbohidratos.
Los monosacáridos son azúcares simples; No se hidrolizan, es decir, que no se descomponen en otros compuestos más simples; Poseen de 3 a 7 átomos de carbono; Su fórmula empírica es (CH2O)n donde n ≥ 3; Se nombran haciendo referencia al número de
carbonos (3-7), terminado en el sufijo -osa.
carbonos (3-7), terminado en el sufijo -osa.
Los monosacáridos más importante son: Glucosa (es la mas principal), Fructosa, Galactosa.
Glucosa:
La glucosa es un monosacárido con fórmula molecular C6H12O6. Es una hexosa, es decir, contiene 6 átomos de carbono, y es una aldosa. Su rendimiento energético es de 4 kilocalorías por cada gramo.
 |
| Glucosa |
Fructosa:
Es un monosacárido con la misma fórmula empírica que la glucosa pero con diferente estructura, es decir, es un isómero de esta. Es una hexosa (6 átomos de carbono), pero cicla en furano (al contrario que las otras hexosas, que lo hacen en pirano)
 |
| Fructosa |
Galactosa:
La galactosa es un monosacárido con fórmula molecular C6H12O6. Es una hexosa, es decir, contiene 6 átomos de carbono, y es una aldosa. A diferencia de la glucosa, la galactosa no se encuentra libre sino que forma parte de la lactosa de la leche. Precisamente es en las glándulas mamarias donde este compuesto se sintetiza para formar parte de la leche materna.Es un isómero de la fructosa, con diferente posición relativa de los grupos -OH y =O
 |
| Galactosa |
Ciclación de aldosas y hexosas:
La causa de una asimetría adicional es una reacción de ciclación intramolecular, que produce un nuevo centro quiral en el átomo de carbono del grupo carbonilo. Esta ciclación se parece a la reacción de un alcohol con un aldehído para formar un hemiacetal, o con una cetona para formar un hemicetal. El carbono carbonílico de una aldosa que contenga al menos cinco átomos de carbono, o de una cetosa que contenga al menos seis átomos de carbono, puede reaccionar con un grupo hidroxilo intramolecular y formar un hemiacetal cíclico o un hemicetal cíclico, respectivamente. El átomo de oxígeno del grupo hidroxilo reaccionante se convierte en miembro de las estructuras anulares con cinco o seis miembros. Como se parece al compuesto heterocíclico pirano, de seis miembros, al anillo con seis miembros de un monosacárido se le llama piranosa. De igual modo, como el anillo con cinco miembros de un monosacárido se parece al del furano, se le llama furanosa. Sin embargo, a diferencia del pirano y del furano, los anillos de los carbohidrato no contienen dobles enlaces.

El carbono más oxidado de un monosacárido ciclado, el que está unido a dos átomos de oxígeno, se llama carbono anomérico. En las estructuras de anillo, el carbono anomérico es quiral. Así, la aldosa o cetosa ciclada puede adoptar cualquiera de dos configuraciones (designadas a o b) para la D-glucosa. A los isómeros a y b se les llama anómeros.
Proyecciones de Haworth:
Es una forma común de representar la fórmula estructural cíclica de los monosacáridos con una perspectiva tridimensional simple. Recibe su nombre del químico inglés sir Walter Norman Haworth. quien trabajó sobre reacciones de ciclación de carbohidratos y propuso esas representaciones. Una proyección de Haworth indica la estereoquímica en forma adecuada, y se puede relacionar con facilidad con una proyección de Fischer. Un monosacárido cíclico se traza de modo que el carbono anomérico esté en la derecha, y los demás carbonos se numeran en dirección de las manecillas del reloj. Los grupos hidroxilo que apuntan hacia abajo en la proyección de Haworth, lo hacen hacia la derecha del esqueleto de carbonos en la proyección de Fischer, en tanto que los grupos hidroxilo que apuntan hacia arriba en la proyección de Haworth lo hacen hacia la izquierda en la proyección de Fischer. En una proyección de Haworth, la configuración del átomo de carbono anomérico se indica con a si su grupo hidroxilo está al lado contrario del anillo respecto del grupo —CH2OH de C-5. Es b si su grupo hidroxilo está en el mismo lado del anillo que el grupo —CH2OH. C-5 es el centro quiral que distingue los azúcares D y L. En las proyecciones de Haworth de los azúcares D, el grupo —CH2OH siempre apunta hacia arriba.

Conformación de los monosacáridos:
Proyección de Haworth son las mas utilizadas en bioquímica. Sin embargo, la geometría de los átomos de carbono en un anillo monosacáridos es tetraédrica (110°). Los monosacáridos cíclicos pueden tener diversas conformaciones, o formas tridimensionales. Los anillos furanosas adoptan conformaciones de sobre o torcida. Los diversos confórmeros de monosacáridos no sustituidos pueden
convertirse rápidamente entre si. Los anillos de piranosa tienden a asumir una de dos conformaciones, la de silla o la de bote.
convertirse rápidamente entre si. Los anillos de piranosa tienden a asumir una de dos conformaciones, la de silla o la de bote.
 |
| Conformación de Bote |
 |
| Conformación de Silla |
Derivados de los monosacáridos:
Estos son:
- Fosfato de azúcar: Los monosacáridos, en las vías metabólicas, con frecuencia
se convierten en esteres de fosfato. - Desoxi y amino azucares: Desoxiazucares ( En estos derivados, un átomo de hidrogeno sustituye a uno de los grupos hidroxilo del monosacárido precursor.). Aminoazucares( En varios azucares, un grupo amino sustituye uno de los gruposhidroxilo del monosacárido precursor. A veces el grupo amino esta acetilado (acetilamino)).
- Azucares alcoholes: En un azúcar alcohol el oxígeno carbonílico del monosacárido precursor se ha reducido y se produce un polihidroxialcohol.
- Azucares ácidos: Los azúcares ácidos son ácidos carboxílicos derivados de las aldosas, sea por oxidación de C-1 (el carbono aldehídico) para formar un ácido aldónico, o por oxidación del carbono con número mayor (el que tiene el alcohol primario) para formar un ácido aldurónico.
- Ácidos ascórbico (vitamina C): El acido L-ascórbico o vitamina C, es un enodiol de una lactona derivada del Dglucoronato. Los primates no pueden convertir glucoronato en acido ascórbico y en consecuencia deben obtenerlos de la dieta. El acido ascórbico es un cofactor esencial para las enzimas que catalizan lahidroxilación de los residuos de prolina y lisina durante la síntesis de colágeno.
Los oligosacáridos son uniones de 2 o mas monosacáridos. El enlace que une dos monosacáridos se llama enlace glicosídico (es el principal enlace estructural en todos los polímeros de los monosacáridos. Es un enlace acetal, donde el carbono anomérico de un azúcar se condensa con un alcohol, una amina o un tiol).
Los oligosacáridos mas Importantes: Sacarosa; Maltosa; Lactosa.
Sacarosa: es un disacárido de glucosa y fructosa. Se sintetiza en plantas, pero no en animales superiores. Es la unión dela glucosa y fructosa.

Lactosa: es un disacárido formado por la unión de una molécula de glucosa y otra de galactosa. No se encuentra en forma natural en las plantas. Se encuentra principalmente en la leche. Es la unión de la Galactosa y Glucosa

Maltosa: Es también conocida como maltobiosa o azúcar de malta, es un disacárido formado por dos glucosas unidas por un enlace glucosídico. Se forma para indicar el germinado de las semillas. Son muy pocos los alimentos que contienen maltosa, por lo que su contribución como fuente de energía al organismo es mínima.Es la unión de la glucosa y glucosa.
Polisacárido:
Con frecuencia se divide a los polisacáridos en dos clases extensas. Los homoglicanos (u homopolisacáridos) son polímeros que sólo contienen residuos de un tipo de monosacárido. Los heteroglicanos (o heteropolisacáridos) son polímeros que contienen residuos de más de un tipo de monosacárido.
Clasificación de los polisacáridos:
Según su composición:
• Homopolisacáridos
• Heteropolisacáridos
Según su función:
• Polisacáridos de reserva: Almidón, Glucógeno.
• Polisacáridos estructurales: Celulosa, Quitina.
Según su composición:
• Homopolisacáridos
• Heteropolisacáridos
Según su función:
• Polisacáridos de reserva: Almidón, Glucógeno.
• Polisacáridos estructurales: Celulosa, Quitina.
Almidón y glucógeno: Son polisacáridos de almacenamiento. Todas las especies sintetizan D-glucosa. Los residuos de glucosa se almacenan como polisacáridos, hasta que se necesiten para producir energía. En plantas y los hongos la forma de almacenar glucosa se conoce como almidón.En animales es el glucógeno.
Celulosa: Es un polisacárido estructural en plantas.
Quitina: Es un polisacárido estructural en el exoesqueleto de muchos artrópodos y moluscos.
Glicoconjugados:
Los glicoconjugados consisten en polisacáridos unidos a (conjugados con) proteínas o péptidos. En muchos casos, los polisacáridos consisten en varias unidades distintas de monosacárido. Por consiguiente, son heteroglicanos. (Almidón, glucógeno, celulosa y quitina son homoglicanos). Los heteroglicanos aparecen en tres tipos de glicoconjugados: proteoglicanos, peptidoglicanos y glicoproteínas.
Se clasifican en:
Proteoglicanos:
Celulosa: Es un polisacárido estructural en plantas.
Quitina: Es un polisacárido estructural en el exoesqueleto de muchos artrópodos y moluscos.
Glicoconjugados:
Los glicoconjugados consisten en polisacáridos unidos a (conjugados con) proteínas o péptidos. En muchos casos, los polisacáridos consisten en varias unidades distintas de monosacárido. Por consiguiente, son heteroglicanos. (Almidón, glucógeno, celulosa y quitina son homoglicanos). Los heteroglicanos aparecen en tres tipos de glicoconjugados: proteoglicanos, peptidoglicanos y glicoproteínas.
Se clasifican en:
- Proteoglicanos
- Peptidoglicanos
- Glicoproteínas
Proteoglicanos:
Son complejos de proteínas y una clase de polisacáridos llamados glicosaminoglicanos. Esos glicoconjugados se presentan principalmente en la matriz extracelular (tejido conectivo) de animales multicelulares. Los glicosaminoglicanos son heteroglicanos no ramificados de unidades repetitivas de disacárido. Como indica el nombre glicosaminoglicano, un componente del disacárido es un amino azúcar, ya sea D-galactosamina (GalN) o D-glucosamina (GlcN).

Peptidoglicanos:
Los peptidoglicanos son polisacáridos unidos a péptidos pequeños. Las paredes celulares de muchas bacterias contienen una clase especial de peptidoglicano con un componente de heteroglicano unido a un péptido de cuatro o cinco residuos. El componente peptídico de los peptidoglicanos varía entre las bacterias. En Staphylococcus aureus es un tetrapéptido con aminoácidos L y D alternados: L-Ala-DIsoglu-L-Lys-D-Ala.
Los peptidoglicanos son polisacáridos unidos a péptidos pequeños. Las paredes celulares de muchas bacterias contienen una clase especial de peptidoglicano con un componente de heteroglicano unido a un péptido de cuatro o cinco residuos. El componente peptídico de los peptidoglicanos varía entre las bacterias. En Staphylococcus aureus es un tetrapéptido con aminoácidos L y D alternados: L-Ala-DIsoglu-L-Lys-D-Ala.
Glicoproteínas
Las glicoproteínas, como los proteoglicanos, son proteínas que contienen oligosacáridos unidos en forma covalente (es decir, proteínas que están glicosiladas; los proteoglicanos son un tipo de glicoproteína). Las cadenas de carbohidrato de una glicoproteína varían de longitud, de 1 hasta más de 30 residuos, y pueden formar hasta 80% de la molécula.
Las glicoproteínas, como los proteoglicanos, son proteínas que contienen oligosacáridos unidos en forma covalente (es decir, proteínas que están glicosiladas; los proteoglicanos son un tipo de glicoproteína). Las cadenas de carbohidrato de una glicoproteína varían de longitud, de 1 hasta más de 30 residuos, y pueden formar hasta 80% de la molécula.
Las cadenas de oligosacárido de las glicoproteínas muestran gran variabilidad de composición. La composición de esas cadenas puede variar, aun entre moléculas de la misma proteína, fenómeno que se llama microheterogeneidad.
Hay varios factores que contribuyen a la diversidad estructural de las cadenas de
oligosacárido en las glicoproteínas.
Hay varios factores que contribuyen a la diversidad estructural de las cadenas de
oligosacárido en las glicoproteínas.
1. Una cadena de oligosacárido puede contener varios azúcares diferentes.
2. Los azúcares pueden estar unidos por enlaces glicosídicos a o b.
3. También los enlaces pueden unir varios átomos de carbono en los azúcares.
3. También los enlaces pueden unir varios átomos de carbono en los azúcares.
4. Las cadenas de oligosacáridos de las glicoproteínas pueden contener hasta cuatro
ramas.
ramas.
Las cadenas de oligosacáridos de la mayor parte de las glicoproteínas están enlazadas a través de O o de N. En los oligosacáridos unidos por O, un residuo de GalNAc se enlaza, en forma típica, a la cadena lateral de un residuo de serina o de treonina. En los oligosacáridos unidos por N, un residuo de GlcNAc está unido al nitrógeno de amida en un residuo de asparagina.
Hay cuatro subclases importantes de enlaces O-glicosídicos en las glicoproteínas.
1. El enlace O-glicosídico más común es el de GalNAc-Ser/Thr mencionado antes. Hay otros azúcares, por ejemplo, galactosa y ácido siálico, que con frecuencia se unen al residuo de GalNAc.
1. El enlace O-glicosídico más común es el de GalNAc-Ser/Thr mencionado antes. Hay otros azúcares, por ejemplo, galactosa y ácido siálico, que con frecuencia se unen al residuo de GalNAc.
2.Algunos de los residuos de 5-hidroxilisina (Hyl) de colágena se unen a la D-galactosa a través de un enlace O-glicosídico. Esta estructura es exclusiva de la colágena.
3. Los glicosaminoglicanos de ciertos proteoglicanos están unidos a la proteína interior mediante una estructura Gal-Gal-Xyl-Ser.
4. En algunas proteínas, un solo residuo de GlcNAc está enlazado a serina o treonina.
Fármacos con Carbohidratos
4. En algunas proteínas, un solo residuo de GlcNAc está enlazado a serina o treonina.
Fármacos con Carbohidratos
Heparina
 Es un glicosaminoglicano muy sulfatado, se utiliza ampliamente cómo anticoagulante inyectable, y tiene la densidad de carga más alta conocida de todas las biomoléculas. También se puede utilizar para formar una superficie interior anticoagulante en diversos dispositivos experimentales y médicos tales como tubos de ensayo y máquinas de diálisis renal.
Es un glicosaminoglicano muy sulfatado, se utiliza ampliamente cómo anticoagulante inyectable, y tiene la densidad de carga más alta conocida de todas las biomoléculas. También se puede utilizar para formar una superficie interior anticoagulante en diversos dispositivos experimentales y médicos tales como tubos de ensayo y máquinas de diálisis renal.  Mecanismo de acción
Mecanismo de acción
La heparina es una sustancia natural que actúan de la sangre que interfiere con el proceso de la coagulación sanguínea. Actúa sobre la trombina, que desempeña un importante papel en la formación del coágulo en la sangre. La heparina clásica ejerce su efecto anticoagulante acelerando la formación de complejos moleculares entre la antitrombina III y los factores II (trombina), IX, X, XI y XII, que quedan inactivados. Tiene particular importancia la acción ejercida sobre la trombina y el factor X.
Eritropoyetina
Es una hormona glicoproteíca cuya función principal, aunque no la única, es la regulación de la producción de glóbulos rojos de la sangre.

Acción
Su acción principal es estimular la citopoyesis, pero la EPO actúa también en la diferenciación de las células de precursor y también estimula en pequeña medida la formación de megacariocitos. El papel parácrino de la eritropoyetina en las gónadas y en el útero todavía no ha sido aclarado.
 La carencia de eritropoyetina ocasiona anemia y como consecuencia los síntomas asociados a ella como debilidad muscular, disminución de la tolerancia al ejercicio físico y mareos.
La carencia de eritropoyetina ocasiona anemia y como consecuencia los síntomas asociados a ella como debilidad muscular, disminución de la tolerancia al ejercicio físico y mareos.Lípidos y Membrana
Los lípidos son compuestos orgánicos insolubles en agua (o sólo poco solubles), que se
encuentran en los sistemas biológicos. Los lípidos tienen gran solubilidad en solventes
orgánicos no polares. Son hidrofóbicos (no polares) o bien son anfipáticos (contienen regiones polares y no polares al mismo tiempo). Los lípidos más simples son los ácidos grasos, y tienen la fórmula general R—COOH, donde R representa una cadena de hidrocarburo.
orgánicos no polares. Son hidrofóbicos (no polares) o bien son anfipáticos (contienen regiones polares y no polares al mismo tiempo). Los lípidos más simples son los ácidos grasos, y tienen la fórmula general R—COOH, donde R representa una cadena de hidrocarburo.
Los ácidos grasos son componentes de muchos tipos más complejos de lípidos, incluyendo los triglicéridos o triacilgliceroles, los glicerofosfolípidos y los esfingolípidos. Los lípidos que contienen grupos fosfato se llaman fosfolípidos y los que tienen grupos esfingosina y carbohidrato a la vez se llaman glicoesfingolípidos. Los esteroides, las vitaminas lipídicas y los terpenos se relacionan con la molécula de isopreno, de cinco carbonos, y por consiguiente se llaman isoprenoides. El nombre terpenos se ha aplicado a todos los isoprenoides, pero en general se restringe a los que existen en las plantas.

Ácidos grasos
 |
| Nomenclatura de los ácidos Grasos |
Los ácidos grasos difieren entre sí en la longitud de sus colas de hidrocarburo, la cantidad de dobles enlaces carbono-carbono, las posiciones de los dobles enlaces en las cadenas y la cantidad de ramificaciones. Son una forma de detergente, porque tienen una larga cola hidrofóbica y una cabeza polar. La cantidad de átomos de carbono en los ácidos grasos más abundantes va de 12 a 20, y casi siempre es par, ya que se sintetizan mediante la adición consecutiva de unidades con dos carbonos. En la nomenclatura común se usan letras griegas para identificar a los átomos de carbono. El carbono adyacente al carbono carboxílico (C-2, en la nomenclatura de IUPAC) se le denomina a, y los demás carbonos tienen las letras b, g, d, e, etcétera.La letra griega w (omega) especifica el átomo de carbono más alejado del grupo carboxilo, cualquiera que sea la longitud de la cola de hidrocarburo. (w es la última letra del alfabeto griego).
 Los ácidos grasos que no contienen dobles enlaces carbono-carbono se llaman saturados, en tanto que los que tienen al menos un doble enlace carbono-carbono se clasifican como no saturados o insaturados. Los ácidos grasos no saturados que sólo tienen un doble enlace carbono-carbono se llaman monoinsaturados, en tanto que los que tienen dos o más se denominan poliinsaturados. La configuración de los dobles enlaces en los ácidos grasos no saturados es cis, en general.
Los ácidos grasos que no contienen dobles enlaces carbono-carbono se llaman saturados, en tanto que los que tienen al menos un doble enlace carbono-carbono se clasifican como no saturados o insaturados. Los ácidos grasos no saturados que sólo tienen un doble enlace carbono-carbono se llaman monoinsaturados, en tanto que los que tienen dos o más se denominan poliinsaturados. La configuración de los dobles enlaces en los ácidos grasos no saturados es cis, en general.
Ejemplo de ácidos grasos:
Las propiedades físicas de los ácidos grasos saturados y no saturados son muy variadas. Típicamente, los ácidos grasos saturados son sólidos céreos a temperatura ambiente (22 °C), en tanto que los ácidos grasos no saturados son líquidos a esta temperatura. La longitud de la cadena de hidrocarburo en un ácido graso, y su grado de insaturación, influyen sobre el punto de fusión
Estructura de los principales ácidos grasos poliinsaturados

Triacilgliceroles o Triglicéridos
Son combustibles metabólicos importantes, en especial en los mamíferos.
La oxidación de los ácidos grasos produce mas energía (37 kJ g-1) que la oxidación de proteínas y carbohidratos (16 kJ g-1 cada uno).
Los ácidos grasos se almacenan en forma de lípidos neutros llamados triacilgliceroles.
Los triacilgliceroles están formados por tres residuos de acilo graso esterificados con glicerina.
Los triacilgliceroles son muy hidrofóbicos. Se almacenan en células en forma anhidra, las moléculas no están solvatadas por el agua.
Son combustibles metabólicos importantes, en especial en los mamíferos.
La oxidación de los ácidos grasos produce mas energía (37 kJ g-1) que la oxidación de proteínas y carbohidratos (16 kJ g-1 cada uno).
Los ácidos grasos se almacenan en forma de lípidos neutros llamados triacilgliceroles.
Los triacilgliceroles están formados por tres residuos de acilo graso esterificados con glicerina.
Los triacilgliceroles son muy hidrofóbicos. Se almacenan en células en forma anhidra, las moléculas no están solvatadas por el agua.
Las grasas y los aceites son mezclas de triacilgliceroles. Pueden ser sólidos (grasas) o líquidos (aceites), dependiendo de sus composiciones de ácidos grasos y de la temperatura. Los triacilgliceroles que sólo contienen grupos acilo graso saturado y de cadena larga tienden a ser sólidos a la temperatura corporal, y los que contienen grupos acilo graso no saturados o de cadena corta, tienden a ser líquidos.
La mayor parte de los lípidos en la dieta humana promedio son triacilgliceroles. Esos lípidos se descomponen en el intestino delgado por acción de las lipasas.
La palmitina contiene tres residuos de acido palmítico.

La trioleina contiene tres residuos de acido oleico.
Glicerofosfolípidos
Son los lípidos mas abundantes en la mayor parte de las membranas
biológicas.
Los glicerofosfolípidos o fosfogliceridos al igual que los triacilgliceroles tienen
un soportarte de glicerol.
Los fosfatidatos están presentes en pequeñas cantidades como intermedios
en la biosíntesis y descomposición de glicerofosfolípidos.
Son los lípidos mas abundantes en la mayor parte de las membranas
biológicas.
Los glicerofosfolípidos o fosfogliceridos al igual que los triacilgliceroles tienen
un soportarte de glicerol.
Los fosfatidatos están presentes en pequeñas cantidades como intermedios
en la biosíntesis y descomposición de glicerofosfolípidos.

Estructura de algunos glicerofosfolipidos: fosfatidilcolina, fosfatidiletanolamina, fosfatidilserina, fosfatidilinositol y difosfotidilglicerol.
La fosfatidilcolina es una familia de licerofosfolipidos y es el fosfolípido más
abundante en las membranas de las células tanto animales como vegetales donde se encuentra en la cara externa de la membrana.
abundante en las membranas de las células tanto animales como vegetales donde se encuentra en la cara externa de la membrana.
También forma parte de las lipoproteínas plasmáticas, es fuente de 1,2-diacilglicerol
(molécula de señalización) mediante la acción de la fosfolipasa D y aporta la fosforilcolina
para la síntesis de esfingomielina.
(molécula de señalización) mediante la acción de la fosfolipasa D y aporta la fosforilcolina
para la síntesis de esfingomielina.
En general, los glicerofosfolipidos tienen ácidos grasos saturados esterificados en el C-1 y
ácidos grasos no saturados esterificados en el C-2.
La fosfatidiletanolamina es también un componente básico de la cara interna de las membranas, en animales y plantas, puede actuar como carabina molecular durante el ensamblaje de las proteínas de membrana guiando su plegamiento y facilitando su transición desde el entorno del citoplasma a la membrana plasmática.
ácidos grasos no saturados esterificados en el C-2.
La fosfatidiletanolamina es también un componente básico de la cara interna de las membranas, en animales y plantas, puede actuar como carabina molecular durante el ensamblaje de las proteínas de membrana guiando su plegamiento y facilitando su transición desde el entorno del citoplasma a la membrana plasmática.
La fosfatidilserina contribuye a las interacciones electrostáticas no
específicas de la cara interna de las membranas aunque esta localización se puede ver alterada durante la activación de plaquetas o en la apoptosis.
Cuando se transfiere a la cara externa de la membrana y actúa como una señal para otras células.
Junto al diacilglicerol y a los iones Ca2+, es necesaria para la activación de la proteína quinasa C (PKC), una enzima clave en la transducción de señales.
específicas de la cara interna de las membranas aunque esta localización se puede ver alterada durante la activación de plaquetas o en la apoptosis.
Cuando se transfiere a la cara externa de la membrana y actúa como una señal para otras células.
Junto al diacilglicerol y a los iones Ca2+, es necesaria para la activación de la proteína quinasa C (PKC), una enzima clave en la transducción de señales.
Para determinar las estructuras de los glicerofosfolípidos y las identidades de sus ácidos grasos individuales se utilizan diversas fosfolipasas que catalizan en forma específica la hidrolisis de los enlaces éster.
La fosfolipasa A2 es la principal enzima en le jugo pancreático y es la responsable de la
digestión de fosfolípidos de membrana la dieta. También esta presente en los venenos de víboras, abejas y avispas.
digestión de fosfolípidos de membrana la dieta. También esta presente en los venenos de víboras, abejas y avispas.
PlasmalogenosOtra clase principal de glicerofosfolípidos, presenta un sustituyente hidrocarburo en el grupo hidroxilo del C-1 de la glicerina unido por un enlace de éter vinílico y no enlace éster.
La etanolamina o la colina se suelen esterificar al grupo fosfato de los plasmalogenos.Estos forman un 23 % de los glicerofosfolípidos en el sistema nervioso central humano y también se encuentran en las membranas de los tejidos nerviosos periféricos y muscular.
La etanolamina o la colina se suelen esterificar al grupo fosfato de los plasmalogenos.Estos forman un 23 % de los glicerofosfolípidos en el sistema nervioso central humano y también se encuentran en las membranas de los tejidos nerviosos periféricos y muscular.
Esfingolípidos
Después de los glicerofosfolípidos, los lípidos mas abundantes en las membranas vegetales y animales son los esfeingolípidos.
Se encuentra en mamíferos en especial abundancia en el tejido nervioso central. La mayor parte de las bacterias no tienen esfingolipiodos.
Después de los glicerofosfolípidos, los lípidos mas abundantes en las membranas vegetales y animales son los esfeingolípidos.
Se encuentra en mamíferos en especial abundancia en el tejido nervioso central. La mayor parte de las bacterias no tienen esfingolipiodos.
El componente estructural de los esfingolipidos es la esfingosina (trans 4- esfingenina). Es un alcohol no ramificado de C 18, con un doble enlace
trans entre el C-4 y C-5 un grupo amino en el C-2 y grupos hidroxilos en el C-1 y C-3.
trans entre el C-4 y C-5 un grupo amino en el C-2 y grupos hidroxilos en el C-1 y C-3.

Ceramida
La ceramida esta formada por un grupo acilo graso unido al grupo amino del C-2 en la esfingosina, por un enlace amida.
Son los precursores metabólicos de todos los esfingolípidos.
Las tres grandes familias de esfingolípidos son las esfingomielinas, los cerebrosidos y los
gangliosidos.
Solo las esfingomielinas contienen fosfato y se clasifican como fosfolípidos, los cerebrocidos y los gangliosidos contienen residuos de carbohidratos y se clasifican como glicoesfingolípidos.
La ceramida esta formada por un grupo acilo graso unido al grupo amino del C-2 en la esfingosina, por un enlace amida.
Son los precursores metabólicos de todos los esfingolípidos.
Las tres grandes familias de esfingolípidos son las esfingomielinas, los cerebrosidos y los
gangliosidos.
Solo las esfingomielinas contienen fosfato y se clasifican como fosfolípidos, los cerebrocidos y los gangliosidos contienen residuos de carbohidratos y se clasifican como glicoesfingolípidos.
Esfingomielina
La fosfocolina esta unida al grupo hidroxilo en el C-1 de una ceramida
Las esfingomielinas existen en las membranas plasmáticas de la mayor parte de las células de mamíferos y son los componentes principales de las vainas de mielina que rodean a ciertas células nerviosas.
La fosfocolina esta unida al grupo hidroxilo en el C-1 de una ceramida
Las esfingomielinas existen en las membranas plasmáticas de la mayor parte de las células de mamíferos y son los componentes principales de las vainas de mielina que rodean a ciertas células nerviosas.
Cerebrosidos
Galactocerebrosiado
Son esfingolípidos que contienen un residuo de monosacárido unido a un enlace β-glicosídico al C-1 de una ceramida.
Los galactocerebrosidos, llamados también galactosilceramidas, tienen un solo residuo de β-D-galactosilo como grupo de cabeza polar.
Los galactocerebrosidos abundan en el tejido nervioso y forman casi el 15% de los lípidos en la vaina de mielina.
Muchos otros tejidos en los mamíferos contienen glucocerebrosidos, ceramidas con un grupo β-D-glucosilo en la cabeza.
Galactocerebrosiado
Son esfingolípidos que contienen un residuo de monosacárido unido a un enlace β-glicosídico al C-1 de una ceramida.
Los galactocerebrosidos, llamados también galactosilceramidas, tienen un solo residuo de β-D-galactosilo como grupo de cabeza polar.
Los galactocerebrosidos abundan en el tejido nervioso y forman casi el 15% de los lípidos en la vaina de mielina.
Muchos otros tejidos en los mamíferos contienen glucocerebrosidos, ceramidas con un grupo β-D-glucosilo en la cabeza.

Gangliosidos
Son glicoesfingolípidos mas complejos, donde las cadenas de oligosacáridos que contienen ácido N-acetilneuraminico (NeuNAc) están unidos a una ceramida.
Gangliosido GM2, la M representa monosialo, es decir un residuo de NeuNAc y el subíndice 2 ya que es el segundo glangliosido que se caracterizo.
Se han caracterizado mas de 60 variedades de gangliosidos.
Se han caracterizado mas de 60 variedades de gangliosidos.
 Esteroides
EsteroidesEs la tercera clase de lípidos que se encuentran en las membranas de los eucariotas y muy rara vez en bacterias.
Los esteroides junto con las vitaminas lipídicas y los terpenos, se clasifican como isoprenoides, por que su molécula se relaciona con le isopreno.
a) Estructura química
b) Esqueleto de carbono
c) Unidad de isopreno
Los esteroides contiene cuatro anillos fundidos,tres de seis carbonos idénticos como A, B y C y un anillo D de cinco carbonos.
La estructura anular característica se deriva del ESCUALENO
Colesterol
El colesterol es un esterol (lípido) que se encuentra en los tejidos corporales y en el plasma sanguíneo de los vertebrados. Se presenta en altas concentraciones en el hígado, médula espinal, páncreas y cerebro.
Pese a tener consecuencias perjudiciales en altas concentraciones, es esencial para crear la membrana plasmática que regula la entrada y salida de sustancias que
atraviesan la célula.
Abundan en las grasas de origen animal.
El colesterol es un esterol (lípido) que se encuentra en los tejidos corporales y en el plasma sanguíneo de los vertebrados. Se presenta en altas concentraciones en el hígado, médula espinal, páncreas y cerebro.
atraviesan la célula.
Abundan en las grasas de origen animal.
El colesterol es un esterol (lípido) que se encuentra en los tejidos corporales y en el plasma sanguíneo de los vertebrados. Se presenta en altas concentraciones en el hígado, médula espinal, páncreas y cerebro.

Otros lípidos de importancia biológica
Hay muchas clases de lípidos que no se encuentran en las membrana.Entre ellos tenemos las ceras, eicosanoides y algunos isoprenoides.
Los lípidos no constituyentes de membranas tienen diversas funciones especializadas (ej. Las vitaminas lipídicas).
Los lípidos no constituyentes de membranas tienen diversas funciones especializadas (ej. Las vitaminas lipídicas).

Ceras
Son ésteres no polares de ácidos grasos de cadena larga y alcoholes monohidroxílicos de cadena larga. Las ceras están muy distribuidas en la naturaleza. Proporcionan cubiertas protectoras impermeables a las hojas y frutos de ciertas plantas, y en la piel, cuero, plumas y exoesqueletos de animales. Por ejemplo, el palmitato de miricilo, uno de los principales componentes de la cera de abejas, es el éster de palmitato (16:0) y del alcohol miricílico, de 30 carbonos (figura 9.17). La hidrofobicidad del palmitato de miricilo hace que la cera de abejas sea muy insoluble en agua, y su alto punto de fusión (debido a las cadenas largas de hidrocarburo saturado) hace que esa cera sea dura y sólida a las temperaturas peculiares de la intemperie.
 |
| Palmitato de miricilo |
Los eicosanoides son derivados oxigenados de ácidos grasos poliinsaturados de C20, como ácido araquidónico. Éstos participan en una diversidad de procesos fisiológicos, y también pueden mediar muchas respuestas potencialmente patológicas.
Las prostaglandinas son eicosanoides que tienen un anillo de ciclopentano. La prostaglandina E2 puede causar constricción de vasos sanguíneos, y el tromboxano A2 interviene en la formación de coágulos sanguíneos, que en algunos casos pueden bloquear el flujo de sangre al corazón o al cerebro. El leucotrieno D4, mediador de la contracción de músculos lisos, también provoca la constricción bronquial de los asmáticos. La aspirina (ácido acetilsalicílico) alivia el dolor, la fiebre, la hinchazón y la inflamación, al inhibir la síntesis de las prostaglandinas.
Las membranas biológicas están formadas por bicapas lipídicas y proteínas
Las membranas biológicas definen los límites externos de las células, y separan compartimientos dentro de ellas. Son componentes esenciales de todas las células vivas. Una membrana típica está formada por dos capas de moléculas de lípidos y muchas proteínas embebidas en ella.
Algunas proteínas contenidas en las membranas sirven como bombas selectivas que controlan en forma estricta el transporte de iones y de moléculas pequeñas que entran y salen de la célula. Las membranas también son responsables de generar y mantener la concentración de gradientes de protones, esenciales para la producción de ATP. Los receptores en las membranas reconocen señales extracelulares y las comunican al interior de la célula.
Bicapas lipídicas
Los glicerofosfolipidos y los glicoesfingolipidos anfipáticas pueden formar monocapas bajo ciertas condiciones lo que los hace ideal para formar bicapa lipídicas.
Las bicapas lipídicas son el principal componente estructural de todas las membranas biológicas, incluyendo membranas plasmáticas y membranas internas de eucariotas.
Las interacciones no covalente entre las moléculas de lípidos en las bicapas hace que la membrana sea flexible y les permite autosellarse.
Las bicapas lipídicas son el principal componente estructural de todas las membranas biológicas, incluyendo membranas plasmáticas y membranas internas de eucariotas.
Las interacciones no covalente entre las moléculas de lípidos en las bicapas hace que la membrana sea flexible y les permite autosellarse.

Modelo fluido de mosaico para membranas biológicas
Una membrana biológica típica contiene de un 25 a un 50% de lípidos, y de un 50 a un 75% de proteínas, en masa, con menos de 10% de carbohidratos como componente de glicolípidos y glicoproteínas. Los lípidos son una mezcla compleja de fosfolípidos, glicoesfingolípidos (en animales) y colesterol (en algunos eucariotas). El colesterol y algunos otros lípidos que por sí no forman bicapas (30% del total) están estabilizados en el arreglo de bicapa por el otro 70% de los lípidos en la membrana. Las composiciones
de las membranas biológicas varían en forma considerable entre las especies, y aun entre distintos tipos celulares en organismos multicelulares.
de las membranas biológicas varían en forma considerable entre las especies, y aun entre distintos tipos celulares en organismos multicelulares.
Una membrana biológica es más gruesa que una bicapa lipídica: en forma típica tiene de 6 a 10 nm de espesor. S. Jonathan Singer y Garth L. Nicolson propusieron en 1972 el modelo de mosaico fluido y todavía tiene validez general para describir el arreglo de lípidos y proteínas dentro de una membrana. Según el modelo del mosaico fluido, la membrana es una estructura dinámica en la que se pueden difundir lateralmente o girar dentro de la bicapa, en forma rápida y aleatoria, las proteínas y los lípidos. Las proteínas de membrana se conciben como témpanos de hielo flotando en un mar muy fluido de bicapa lipídica.

Las bicapas lipídicas y las membranas son estructuras dinámicas
Los lípidos en una bicapa están en movimiento constante, dando a las bicapas lipídicas muchas de las propiedades de los fluidos. Los lípidos tienen varios tipos de movimiento molecular dentro de las bicapas:
a) Difusión lateral, es relativamente rápida y el movimiento de los lípidos dentro del plano de una monocapa. Es bidimensional.
b) Difusión transversal o basculante de los lípidos es muy lenta y es el paso de lípidos de una monocapa de la bicapa a la otra.
b) Difusión transversal o basculante de los lípidos es muy lenta y es el paso de lípidos de una monocapa de la bicapa a la otra.

Todas las células sintetizan membranas nuevas agregando lípidos y proteínas a sus membranas existente.
Al extenderse la membrana plasmática, la célula aumenta de tamaño.
Al extenderse la membrana plasmática, la célula aumenta de tamaño.
Al final la célula se dividirá y cada célula hija hereda una parte (por la mitad) de la membrana progenitora.
Difusión de proteínas en membranas
1970, L. D. Frye y Michael A., realizaron experimentos para probar si las proteínas de membrana se difunden dentro de la bicapa lipídica. Con microscopia de inmunofluorescencia, observaron que las proteínas marcadas se entrecruzaron dentro de 40 minutos después de la fusión celular. Al menos algunas proteínas de membrana se difunden libremente en las membranas biológicas.

Tres clases de proteínas de membrana
Las membranas celulares e intracelulares contienen proteínas
especializadas enlazadas en la membrana.
1. Proteínas integrales de membrana.
2. Proteínas especificas de membrana.
3. Proteínas de membranas ancladas a lípidos.
Según su modelo de asociación con la bicapa
lipídica:
Las membranas celulares e intracelulares contienen proteínas
especializadas enlazadas en la membrana.
1. Proteínas integrales de membrana.
2. Proteínas especificas de membrana.
3. Proteínas de membranas ancladas a lípidos.
Según su modelo de asociación con la bicapa
lipídica:
1. Proteínas integrales de membrana:
Estas proteínas se caracterizan por tener regione hidrofóbicas incrustadas en el interior hidrofóbico de la bicapa lipídica. Abarcan totalmente la bicapa, con una parte de la proteína expuesta sobre la superficie externa o otra expuesta sobre la superficie interna.

2. Proteínas especificas de membrana.
Se asocian a una cara de la membrana mediante interacciones de carga – carga y con puentes de hidrogeno, con las proteínas integrales de membrana o con los grupos de cabeza polar de los lípidos de membrana.

3. Proteínas de membranas ancladas a lípidos.
Están unidas a una membrana mediante un enlace covalente con un ancla lipídico.
Los tres tipos de anclas se pueden encontrar en la misma membrana, pero no forman un complejo.
a) Proteína anclada a un acilo graso.
b) Proteína de membrana anclada a un prenilo. Las proteínas de membranas ancladas en acilo graso y prenilo también existen en la hojilla citoplasmática (externa) de la membranas intracelulares.
c) Proteína anclada por glicosilfosfatidilinositol.
a) Proteína anclada a un acilo graso.
b) Proteína de membrana anclada a un prenilo. Las proteínas de membranas ancladas en acilo graso y prenilo también existen en la hojilla citoplasmática (externa) de la membranas intracelulares.
c) Proteína anclada por glicosilfosfatidilinositol.

Transporte de membrana
A.Termodinámica del transporte en la membrana
B.Poros y canales
C.Transporte pasivo
D.Transporte activo
E.Endocitosis y exocitosis
A.Termodinámica del transporte en la membrana
B.Poros y canales
C.Transporte pasivo
D.Transporte activo
E.Endocitosis y exocitosis
A.Termodinámica del transporte en la membrana
Para una molécula A, la concentración en el interior de la membrana es [Aint] y la concentración en el exterior es [Aext]. El cambio de energía libre de Gibbs asociado al transporte de moléculas de A es:

Si la concentración de A en el interior de la célula es mucho menor que fuera de la célula, entonces ΔG transporte será negativa y se favorecerá termodinámicamente la entrada de A a la célula. Por ejemplo, si [Aint] = 1 mM y [Aext] = 100 mM, entonces, a 25 °:

B. Poros y canales
Los poros y los canales son proteínas transmembranales (porinas) con un paso central para iones y moléculas pequeñas. (En general, el término poro se usa en las bacterias, y canal para los animales). En este proceso no se requiere energía. Se controla por difusión.
Transporte de membrana a través de un poro o un canal. Un pasaje central permite que las moléculas y iones del tamaño, carga y geometría adecuados atraviesen la membrana en cualquier dirección.
C.Transporte pasivo:
El transporte pasivo también se llama difusión facilitada, porque no requiere fuente de energía. La proteína de transporte acelera el movimiento del soluto a favor de su
gradiente de concentración. Las proteínas de transporte son parecidas a las enzimas, porque aumentan la velocidad de un proceso que es termodinámicamente favorable. La ecuación que describe esta dependencia se parece a la ecuación de Michaelis-Menten para catálisis enzimática.
gradiente de concentración. Las proteínas de transporte son parecidas a las enzimas, porque aumentan la velocidad de un proceso que es termodinámicamente favorable. La ecuación que describe esta dependencia se parece a la ecuación de Michaelis-Menten para catálisis enzimática.
Cinética del transporte pasivo. La velocidad inicial de transporte aumenta con la concentración del sustrato, hasta que se alcanza un máximo. Ktr es la concentración del sustrato a la cual la velocidad de transporte es la mitad de la máxima.
Tipos de transporte activo y pasivo
Los transportadores de membrana más simples, sean activos o pasivos, realizan uniporte; esto es, sólo llevan un solo tipo de soluto a través de la membrana . Muchos transportadores hacen el transporte simultáneo de dos moléculas de diferentes solutos. Si ambas moléculas se transportan en la misma dirección, el proceso se llama simporte. Si se transportan en direcciones opuestas, el proceso es antiporte
D.Transporte activo:
El transporte activo se parece al transporte pasivo en el mecanismo y propiedades cinéticas generales. En el transporte activo, el soluto se mueve contra un gradiente de concentración, contra una diferencia de carga, o ambas cosas. El transporte activo debe acoplarse a una reacción productora de energía para contrarrestar el cambio desfavorable de energía libre de Gibbs, para transporte sin ayuda.
El transporte activo primario está activado por una fuente directa de energía, como ATP o luz. Por ejemplo, la bacteriorrodopsina usa energía luminosa para generar un gradiente de concentración de protones a través de la membrana, que se puede usar para formación de ATP. El transporte activo secundario está impulsado por un gradiente de concentración. El transporte cuesta arriba activo de un soluto se acopla con el transporte cuesta abajo de un segundo soluto que estaba concentrado por el transporte activo primario.
El transporte activo primario está activado por una fuente directa de energía, como ATP o luz. Por ejemplo, la bacteriorrodopsina usa energía luminosa para generar un gradiente de concentración de protones a través de la membrana, que se puede usar para formación de ATP. El transporte activo secundario está impulsado por un gradiente de concentración. El transporte cuesta arriba activo de un soluto se acopla con el transporte cuesta abajo de un segundo soluto que estaba concentrado por el transporte activo primario.

E.Endocitosis y exocitosis
En las células eucariotas, aunque no en todas, las proteínas (y ciertas sustancias grandes) se mueven hacia adentro y afuera de la célula por endocitosis y exocitosis, respectivamente.
La endocitosis es el proceso mediante el cual las macromoléculas son rodeadas por la membrana plasmática, y son llevadas al interior de la célula dentro de una vesícula lipídica.
La exocitosis se parece a la endocitosis, pero la dirección del transporte es la inversa. Durante la exocitosis, los materiales destinados a ser secretados de la célula se encierran en vesículas mediante el aparato de Golgi. A continuación las vesículas se funden con la membrana plasmática y liberan su contenido al espacio extracelular.
La endocitosis es el proceso mediante el cual las macromoléculas son rodeadas por la membrana plasmática, y son llevadas al interior de la célula dentro de una vesícula lipídica.
La exocitosis se parece a la endocitosis, pero la dirección del transporte es la inversa. Durante la exocitosis, los materiales destinados a ser secretados de la célula se encierran en vesículas mediante el aparato de Golgi. A continuación las vesículas se funden con la membrana plasmática y liberan su contenido al espacio extracelular.

Fármacos relacionados con lípidos
Colestiramina (Questran)
Es una resina polimérica con capacidad para fijar los ácidos biliares. Inicialmente, se utilizó para tratar el prurito secundario a la colestasis, pero hoy día su principal indicación es el tratamiento de la hipercolesterolemia con hipertrigliceridemia concomitante. También se ha utilizado esta resina para tratar la enterocolitis producida por el Clostridium difficile

Dosis:
Administración oral:
- Adultos: inicialmente 4 g de colestiramina tres veces al día, antes de las comidas. Las dosis de deben ajustar de acuerdo con la respuesta clínica. La dosis de mantenimiento más usual es de 4 a 8 g de la resina, 2 a 4 veces al día, con las comidas y a la hora de acostarse. La dosis máxima recomendada es de 24 g/día como hipocolesterolemiante y de 16 g/día como antiprurítico
- Adolescentes: la dosis de 8 g/día ha mostrado ser eficaz y segura en esta población. La dosis de debe repartir en dos tomas, antes de las comidas
- Niños de 6 a 12 años: se recomienda 80 mg/kg por vía oral o 2.35 g/m2 tres veces al día con las comidas. Alternativamente pueden administrarse inicialmente de 2 a 4 g dos veces al día antes de las comidas. Estas dosis se pueden incrementar hasta conseguir la respuesta clínica adecuada o hasta el desarrollo de efectos adversos intolerables, hasta un máximo de 8 g/día
- Niños de < 6 años: la seguridad y eficacia de la colestiramina no han sido evaluados en esta población

Mecanismo de acción
Actúa uniéndose a los ácidos biliares e impidiendo su reabsorción. De esta manera se fomenta la transformación del colesterol hepático en ácidos biliares. Secundariamente, la disminución del colesterol incrementa la actividad de los receptores LDL de los hepatocitos, con lo que se incrementa la eliminación del colesterol LDL plasmático.
Lovastatina (Mevacor)
Es un fármaco miembro de la familia de las estatinas, usado para disminuir el colesterol y prevenir enfermedades cardiovasculares.
Vías de administración (formas de uso)
Oral.
Dosis
En adultos:
- 20 mg/día vía oral inicialmente, como dosis única con la cena, aumentándolo en intervalos de 4 semanas o más hasta una dosis de mantenimiento de 20-80 mg/día en dosis únicas o divididas. Los pacientes con niveles de colesterol por encima de 300 mg/dL (7,8 mmol/L) con dieta como terapia pueden ser iniciados con 40 mg/día.
Absorción
La absorción, al igual que la mayoría de las estatinas, es limitada, reduciéndose a un máximo del 30%. Sufre efecto de primer paso metabólico. Esto junto con la escasa absorción le da una biodisponibilidad muy baja, en torno al 5%. Diversos estudios muestran que su absorción en ayunas es de 2/3 a la registrada inmediatamente después de las comidas.
Mecanismo de acción
El β-hidroxiácido de lovastatina es un inhibidor de la 3-hidroxi-3-metilglutaril-coenzima A reductasa (HMG-CoA reductasa). Esta enzima cataliza un paso esencial de la vía del mevalonato, la conversión de la HMG-CoA amevalonato, que es un metabolito clave en la en la biosíntesis de colesterol. En el esquema adjunto puede observarse el nivel de bloqueo de las estatinas así como de otras sustancias en la biosíntesis del colesterol. La inhibición de la enzima se realiza de forma competitiva. El bloqueo de la síntesis hepática del colesterol produce una activación de las proteínas reguladoras SREBP (sterol regulatory elements-binding proteins), que activan la transcripción de proteínas y, por tanto, producen una mayor expresión del gen del receptor de LDL y un aumento en la cantidad de receptores funcionales en el hepatocito.

Dosis
20-40 mg/día, por la noche, ajuste de dosis cada 4 sem; máx. 80 mg/día. Niños > 9 años: 20-40 mg/día, por la noche, ajuste de dosis cada 6 sem; máx. 80 mg. Enf. cardiaca coronaria después de intervención coronaria: 80 mg/día.
Mecanismo de acción
La fluvastatina interfiere con la actividad de la hidroximetilglutaril-coenzima A reductasa (HMG-CoA), una enzima hepática, con lo que se interrumpe la vía sintética de la biosíntesis de colesterol en el hombre. Como consecuencia, los niveles del colesterol hepático son reducidos, estimulándose la captación de las LDLs. La fluvastatina reduce el colesterol total circulante, el colesterol asociado a las LDLs y los triglicéridos. La fluvastatina es más potente que la lovastatina como inhibidor de la HMG-CoA reductasa.
Ácidos Nucleícos
Son grandes polímeros formados por la repetición de monómeros denominados nucleótido, unidos mediante enlaces fosfodiéster. Se forman, largas cadenas; algunas moléculas de ácidos nucleicos llegan a alcanzar tamaños gigantescos, con millones de nucleótidos encadenados.
Importancia biológica de los ácidos nucleicos
Un organismo vivo contiene un conjunto de instrucciones para formar una réplica de sí mismo.
El genoma del organismo o material genético es donde está toda esa información.
Los genomas de todas las células están formados por ADN. Algunos genomas virales están formados por ARN.
En muchas bacterias, el genoma puede consistir en una solo molécula de ADN.
La información que especifica la estructura primaria de una proteína esta codificada en la secuencia de nucleótidos en el ADN.
Los ácidos nucleícos representan la cuarta gran clase de macromoléculas.
Estas al igual que las proteínas y los polisacáridos, contienen múltiples unidades manométricas similares que se unen en forma covalente para producir polímeros grandes.
El genoma del organismo o material genético es donde está toda esa información.
Los genomas de todas las células están formados por ADN. Algunos genomas virales están formados por ARN.
En muchas bacterias, el genoma puede consistir en una solo molécula de ADN.
La información que especifica la estructura primaria de una proteína esta codificada en la secuencia de nucleótidos en el ADN.
Los ácidos nucleícos representan la cuarta gran clase de macromoléculas.
Estas al igual que las proteínas y los polisacáridos, contienen múltiples unidades manométricas similares que se unen en forma covalente para producir polímeros grandes.
Funciones de los ácidos nucleícos
Duplicación del ADN
Transcripción del ADN para formar ARNm y otros ARN
Traducción, en los ribosomas, del mensaje contenido en el ARNm a proteínas.
Expresión del mensaje genético, proteínas.
Transcripción del ADN para formar ARNm y otros ARN
Traducción, en los ribosomas, del mensaje contenido en el ARNm a proteínas.
Expresión del mensaje genético, proteínas.
En 1869, Friedrich Miescher descubre la sustancia que resulto ser acido desoxirribonucleico (ADN). Acido desoxirribonucleico (ADN) está conformado por
carbono, hidrógeno, oxígeno y alto contenido de fósforo.
El nombre inicial de esta sustancia fue “nucleína”, luego se cambio a acido nucleíco.
Hoppe-Seyler aisló una sustancia muy parecida en las células de levadura, esta sustancia era ARN. 1944, Oswald Avery, Colin MacLeod y Maclyn McCarty demostraron que el ADN es la molécula que contiene la información genética. Por aquella época se conocía muy poco sobre la estructura de ADN.
carbono, hidrógeno, oxígeno y alto contenido de fósforo.
El nombre inicial de esta sustancia fue “nucleína”, luego se cambio a acido nucleíco.
Hoppe-Seyler aisló una sustancia muy parecida en las células de levadura, esta sustancia era ARN. 1944, Oswald Avery, Colin MacLeod y Maclyn McCarty demostraron que el ADN es la molécula que contiene la información genética. Por aquella época se conocía muy poco sobre la estructura de ADN.
Determinación de la estructura del ADN
 El 28 de febrero de 1953 los científicos James Watson y Francis Crick determinan la estructura del ADN. Los científicos propusieron el modelo de la doble hélice de ADN para representar la estructura tridimensional del polímero.
El 28 de febrero de 1953 los científicos James Watson y Francis Crick determinan la estructura del ADN. Los científicos propusieron el modelo de la doble hélice de ADN para representar la estructura tridimensional del polímero.En una serie de cinco artículos en el mismo número de Nature se publicó la evidencia experimental que apoyaba el modelo de Watson y Crick.
De éstos, el artículo de Franklin y Raymond Gosling fue la primera publicación con datos de difracción de rayos X que apoyaba el modelo de Watson y Crick, y en ese mismo número de Nature también aparecía un artículo sobre la estructura del ADN de Maurice Wilkins y sus colaboradores.
Watson, Crick y Wilkins recibieron conjuntamente, en 1962, después de la muerte de Rosalind Franklin, el Premio Nobel en Fisiología o Medicina. Sin embargo, el debate continúa sobre quién debería recibir crédito por el descubrimiento.
Niveles estructurales de los ácidos nucleicos
Estructura secundaria del ADN

Estructura terciaria del ADN
superenrollamiento de la molécula

Estructura cuaternaria del ADN
superenrollamiento de la molécula con otras moléculas

Los nucleótidos son los bloques de construcción de los ácidos nucleicos
Los ácidos nucleicos son polinucleótidos, o polímeros de nucleótidos. Los nucleótidos tienen tres componentes: un azúcar de cinco carbono, uno o más grupos fosfato y un compuesto nitrogenado débilmente básico llamado base. Las bases que se encuentran en los nucleótidos son PIRIMIDINAS y PURINAS sustituidas.
La pentosa suele ser ribosa (D-ribofuranosa) o 2-desoxirribosa (2-desoxi-D-ribofuranosa). Los N-glicósidos pirimidina o purina de estos azúcares se llaman nucleósidos. Los nucleótidos son los ésteres de fosfato de los nucleósidos; los nucleótidos comunes contienen uno a tres grupos fosforilo. Los nucleótidos que contienen ribosa se llaman ribonucleótidos, y los que contienen desoxirribosa se llaman desoxirribonucleótidos. A. Ribosa y desoxirribosa:
Los azúcares componentes de los nucleótidos que se encuentran en los ácidos nucleicos. Los dos azúcares aparecen como proyecciones de Haworth de la configuración b de las formas de anillo de furanosa. Es la configuración estable que existe en los nucleótidos y polinucleótidos. Cada uno de esos anillos de furanosa puede adoptar conformaciones diferentes. La conformación de la desoxirribosa predomina en el ADN de doble hebra.
B. Pirimidinas y purinas
Las bases que se encuentran en los nucleótidos son derivados de pirimidina o de purina.
Estructura de las purinas y pirimidinas
La pirimidina tiene un solo anillo de cuatro átomos de carbono y dos de nitrógeno.La purina tiene un sistema de anillos fundidos de pirimidina y de imidazol. Los dos tiposde bases son no saturados, con dobles enlaces conjugados. Esta propiedad hace que los anillos sean planos, y también explica su capacidad de absorber la luz ultravioleta.
Clasifiación
Las purinas y pirimidinas sustituidas son ubicuas en las células vivas, pero casi nunca se encuentran las bases no sustituidas en los sistemas biológicos. Las principales pirimidinas que hay en los nucleótidos son uracilo (2,4-dioxopirimidina, U), timina
(2,4-dioxo-5-metilpirimidina, T) y citosina (2-oxo-4-aminopirimidina, C). Las principales purinas son adenina (6-aminopurina, A) y guanina (2-amino-6-oxopurina, G). Las estructuras químicas de esas cinco bases principales la timina es una forma sustituida de uracilo, también se puede llamar 5-metiluracilo. La adenina, la guanina y la citosina están en ribonucleótidos y desoxirribonucleótidos. El uracilo se encuentra principalmente en ribonucleótidos y la timina en desoxirribonucleótidos.
(2,4-dioxo-5-metilpirimidina, T) y citosina (2-oxo-4-aminopirimidina, C). Las principales purinas son adenina (6-aminopurina, A) y guanina (2-amino-6-oxopurina, G). Las estructuras químicas de esas cinco bases principales la timina es una forma sustituida de uracilo, también se puede llamar 5-metiluracilo. La adenina, la guanina y la citosina están en ribonucleótidos y desoxirribonucleótidos. El uracilo se encuentra principalmente en ribonucleótidos y la timina en desoxirribonucleótidos.
Las purinas y las pirimidinas son bases débiles relativamente insolubles en agua al pH fisiológico.
Estructura de algunas nucleobases menos comunes
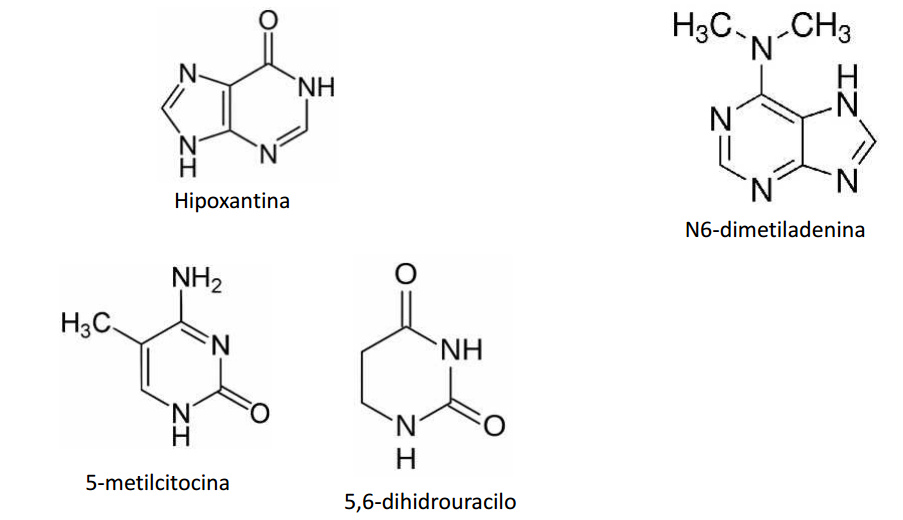
Sin embargo, dentro de las células la mayor parte de bases pirimidina y purina se encuentran como constituyentes de nucleótidos y polinucleótidos, compuestos que son muy hidrosolubles.
Cada base heterocíclica de los nucleótidos comunes puede existir cuando menos en dos formas tautómeras. La adenina y la citosina (que son amidinas cíclicas) pueden existir en sus formas amino o imino, y la guanina, timina y uracilo (que son amidas cíclicas) pueden existir en forma de lactama (ceto) o de lactima (enol). Las formas tautómeras de cada base existen en equilibrio, pero los tautómeros amino y lactama son más estables, y en consecuencia predominan bajo las condiciones que hay en el interior de la mayoría de las células. Los anillos permanecen no saturados y planos en cada tautómero.
Cada base heterocíclica de los nucleótidos comunes puede existir cuando menos en dos formas tautómeras. La adenina y la citosina (que son amidinas cíclicas) pueden existir en sus formas amino o imino, y la guanina, timina y uracilo (que son amidas cíclicas) pueden existir en forma de lactama (ceto) o de lactima (enol). Las formas tautómeras de cada base existen en equilibrio, pero los tautómeros amino y lactama son más estables, y en consecuencia predominan bajo las condiciones que hay en el interior de la mayoría de las células. Los anillos permanecen no saturados y planos en cada tautómero.
Estructuras de los tautómeros

C. Nucleósidos
Los nucleósidos están formados por ribosa y desoxirribosa y una base heterocíclica. En cada nucleósido, un enlace b-N-glicosídico conecta el C-1 del azúcar al N-1 de la pirimidina o al N-9 de la purina. Por consiguiente, los nucleósidos son derivados N-ribosilo o N-desoxirribosilo de las pirimidinas o las purinas. La convención de numeración para los átomos de carbono y nitrógeno de los nucleósidos refleja que están formados por una base y un azúcar de cinco carbonos, y cada uno de ellos tiene su propio esquema de numeración. La designación de los átomos en las partes de purina y pirimidina tiene preferencia. Por consiguiente, los átomos de las bases se numeran 1, 2, 3, etc., en tanto que los del anillo de furanosa se diferencian por tener primas (´ ). Así, el enlace b-N-glicosídico se une al átomo de C-1 , o 1 , de la parte del azúcar a la base. La ribosa y la desoxirribosa difieren en la posición del C-2 , o 2 .
Los nucleósidos están formados por ribosa y desoxirribosa y una base heterocíclica. En cada nucleósido, un enlace b-N-glicosídico conecta el C-1 del azúcar al N-1 de la pirimidina o al N-9 de la purina. Por consiguiente, los nucleósidos son derivados N-ribosilo o N-desoxirribosilo de las pirimidinas o las purinas. La convención de numeración para los átomos de carbono y nitrógeno de los nucleósidos refleja que están formados por una base y un azúcar de cinco carbonos, y cada uno de ellos tiene su propio esquema de numeración. La designación de los átomos en las partes de purina y pirimidina tiene preferencia. Por consiguiente, los átomos de las bases se numeran 1, 2, 3, etc., en tanto que los del anillo de furanosa se diferencian por tener primas (´ ). Así, el enlace b-N-glicosídico se une al átomo de C-1 , o 1 , de la parte del azúcar a la base. La ribosa y la desoxirribosa difieren en la posición del C-2 , o 2 .
 |
| Sitios para enlaces de hidrógeno en las bases de ácidos nucleicos. |
Estructura química de los nucleósidos
 |
| a. Ribonucleósidos y b. Desoxirribonucleósidos |
Conformación sin y anti Adenosina
Algunos nucleótidos toman la conformación sin o anti. En los nucleósidos comunes de pirimidinas predomina la conformación anti. En los ácidos nucleicos, que son polímeros de los nucleótidos, predomina las conformaciones anti.

D. Nucleótidos
Los nucleótidos son derivados fosforilados de los nucleósidos. Los ribonucleósidos contienen tres grupos hidroxilo que se pueden fosforilar (2 , 3 y 5 ), y los desoxirribonucleósidos contienen dos de esos grupos hidroxilo (3 y 5 ). En los nucleótidos naturales, los grupos fosforilo suelen estar unidos al átomo de oxígeno del grupo 5 -hidroxilo. Por convención, siempre se supone que un nucleótido es un éster de 5 -fosfato, a menos que se indique otra cosa.
Estructura química de los desoxirribonucleotido-5’-monofosfatos


Características del ADN
El ADN tiene doble hebra
1950, el ADN es un polímero lineal de residuos de 2’ desoxirribonucleotido unidos por 3’,5’-fosfodiester.Erwin Chargaff había deducido ciertas regularidades en las composiciones de nucleótidos de muestras de ADN obtenidas de gran variedad de procariotas y eucariotas. Entre otras cosas, Chargaff observó que en el ADN de determinada célula están presentes A y T en cantidades equimolares, así como G y C.
El modelo de ADN propuesto por Watson y Crick en 1953 se basó en las estructuras conocidas de los nucleósidos, sobre figuras de difracción de rayos X que obtuvieron Rosalind Franklin y Maurice Wilkins de fibras de ADN, y en las equivalencias químicas notadas por Chargaff. El modelo de Watson-Crick explicó las cantidades iguales de purinas y pirimidinas al sugerir que el ADN tiene doble hebra (doble cadena) y que las bases en una hebra se apareaban en forma específica con las bases de la otra: A con T y G con C. La estructura propuesta por Watson y Crick se llama hoy conformación B del ADN, o simplemente B-ADN.
Es importante apreciar la estructura del ADN para comprender los procesos de su replicación y transcripción. El ADN es el almacén de la información biológica.Cada célula contiene docenas de enzimas y proteínas que se unen al ADN y reconocen ciertas propiedades estructurales, como la secuencia de nucleótidos.
Doble hebra de ADN
A. Unión de nucleótidos por enlaces de 3’,5’ fosfodiester.
B. Formación de una doble hélice con dos hebras antiparalelas.
C. Estabilización de la doble hélice por fuerzas débiles.
D.Conformación de ADN de doble hebra
B. Formación de una doble hélice con dos hebras antiparalelas.
C. Estabilización de la doble hélice por fuerzas débiles.
D.Conformación de ADN de doble hebra
A. Unión de nucleótidos por enlaces de 3’,5’ fosfodiester.
La estructura primaria de una proteína se refiere a la secuencia de sus residuos de aminoácido unidos por enlaces peptídicos; en forma parecida, la estructura primaria de un ácido nucleico es la secuencia de sus residuos de nucleótido unidos por enlaces 3 ,5 -fosfodiéster. Un tetranucleótido tiene Estructura química:pdApdGpdTpdC. Los residuos de nucleótido están unidos por enlaces 3’-5’-fosfodiester.El nucleótido con un grupo 5’-fosforilo libre se llama extremo 5’, y el nucleótido con un grupo 3’-hidroxilo libre se llama extremo 3’.


B. Formación de una doble hélice con dos hebras antiparalelas.
La mayor parte de las moléculas de ADN consisten en dos hebras, de polinucleótidos. Cada una de las bases en una hebra forma puentes de hidrógeno con una base de la hebra opuesta.Los pares de bases mas comunes están entre los tautómeros lactoma y amino de las bases.

La molécula de ADN se puede visualizar como una “escalera” que se ha torcido para formar una hélice. Las bases apareadas representan los peldaños de la escalera, y los esqueletos de azúcar-fosfato representan los soportes. Cada hebra complementaria sirve como una plantilla perfecta a la otra. Esta complementariedad es responsable de la regularidad general de la estructura del ADN de doble hebra. Sin embargo, el apareamiento de bases complementarias solo no produce una hélice. En el B-ADN, los pares de bases se apilan uno sobre otro, y son casi perpendiculares al eje longitudinal de la molécula. Las interacciones no covalentes cooperativas entre las superficies superior e inferior de cada par de base acercan entre sí a esos pares y crean un interior hidrofóbico que hace que se tuerza el esqueleto de azúcar-fosfato.

La doble hélice tiene dos surcos de ancho desigual, por la forma en que se apilan los pares de bases y en que se tuercen los esqueletos de azúcar-fosfato. Esos surcos se llaman surco mayor y surco menor. Dentro de cada surco, los grupos funcionales en las orillas de los pares de bases quedan expuestos al agua. Cada par de bases tiene una pauta distintiva de grupos químicos en los surcos. Como los pares de bases son accesibles en los surcos, las moléculas que interactúan con determinados pares de bases pueden identificarlos sin alterar la hélice. Esto tiene importancia particular en las proteínas que deben unirse al ADN de doble hebra y “leer” determinada secuencia. El B-ADN es una hélice derecha con 2.37 nm de diámetro. El ascenso de la hélice es la distancia entre un par de bases y el siguiente, a lo largo del eje de la hélice, y es de 0.33 nm en promedio; el paso de la hélice es la distancia para completar una vuelta, aproximadamente 3.40 nm. Esos valores varían dentro de ciertos límites que dependen de la composición en bases. Ya que hay unos 10.4 pares de bases por vuelta de la hélice, el ángulo de rotación entre nucleótidos adyacentes en cada hebra es de unos 34.6° (360/10.4).

La longitud de las moléculas de ADN de doble hebra se expresa con frecuencia en términos de pares de bases (pb). Por comodidad, las estructuras más largas se miden en miles de pares de bases o kilopares de bases, que se abrevian kb. La mayor parte de los genomas bacterianos consisten en una sola molécula de ADN de varios miles de kb; por ejemplo, el cromosoma de Escherichia coli tiene 4 600 kb. Las moléculas más grandes de ADN en los cromosomas de mamíferos y plantas de floración pueden ser de varios cientos de miles de kb de longitud. El genoma humano contiene 3 200 000 kb (3.2 109 pares de bases) de ADN.
 |
| a) Modelo de bolas y palillos. Los pares de bases son casi perpendiculares a las columnas vertebrales de azúcar-fosfato.b) Modelo de relleno espacial. Clave de colores: carbono gris, nitrógeno azul, oxígeno rojo, fósforo púrpura. |
C. Estabilización de la doble hélice por fuerzas débiles.
Las fuerzas que mantienen las conformaciones nativas de las estructuras celulares complejas tienen la fuerza suficiente para mantener las estructuras, pero la debilidad suficiente para permitir que haya flexibilidad de conformación. Los enlaces covalentes
entre los residuos adyacentes determinan las formas tridimensionales de esas macromoléculas. Hay cuatro clases de interacciones que afectan la conformación del ADN de doble hebra:
1. Interacciones de apilamiento: Los pares de bases apilados forman contactos de van der Waals. Aunque las fuerzas entre los pares de bases individuales apilados son débiles, son aditivas, por lo que en las moléculas grandes de ADN los contactos de van der Waals son una fuente importante de estabilidad.
2. Puentes de hidrógeno: Los puentes de hidrógeno entre pares de bases forman una importante fuerza estabilizadora.
3. Efectos hidrofóbicos: Al sepultar los anillos hidrofóbicos de purina y pirimidina en el interior de la doble hélice aumenta la estabilidad de la hélice.
4. Interacciones entre cargas: La repulsión electrostática de los grupos fosfato con carga negativa en el esqueleto es una fuente potencial de inestabilidad de la hélice de ADN. Sin embargo, la repulsión se minimiza por la presencia de cationes como y proteínas catiónicas (que contienen abundancia de los residuos básicos arginina y lisina).
entre los residuos adyacentes determinan las formas tridimensionales de esas macromoléculas. Hay cuatro clases de interacciones que afectan la conformación del ADN de doble hebra:
1. Interacciones de apilamiento: Los pares de bases apilados forman contactos de van der Waals. Aunque las fuerzas entre los pares de bases individuales apilados son débiles, son aditivas, por lo que en las moléculas grandes de ADN los contactos de van der Waals son una fuente importante de estabilidad.
2. Puentes de hidrógeno: Los puentes de hidrógeno entre pares de bases forman una importante fuerza estabilizadora.
3. Efectos hidrofóbicos: Al sepultar los anillos hidrofóbicos de purina y pirimidina en el interior de la doble hélice aumenta la estabilidad de la hélice.
4. Interacciones entre cargas: La repulsión electrostática de los grupos fosfato con carga negativa en el esqueleto es una fuente potencial de inestabilidad de la hélice de ADN. Sin embargo, la repulsión se minimiza por la presencia de cationes como y proteínas catiónicas (que contienen abundancia de los residuos básicos arginina y lisina).
El ADN de doble hebra es termodinámicamente más estable que las hebras separadas, lo cual explica por qué predomina la forma de doblehebra in vivo. Sin embargo, a veces se puede alterar la estructura de regiones localizadas de la doble hélice al desenrollarse. Esa alteración sucede durante la replicación, reparación, recombinación y transcripción del ADN. Al desenrollamiento y la separación completos de las hebras individuales complementarias se le llama desnaturalización.La desnaturalización sólo sucede in vitro. El ADN de doble hebra se puede desnaturalizar con calor o con un agentecaotrópico, como urea o cloruro de guanidinio. En estudios de desnaturalización térmica se aumenta lentamente la temperatura de una solución de ADN. Al elevar la temperatura, se dispersan cada vez más bases y se rompen más puentes de hidrógeno entre pares de bases. Llega un momento en que las dos cadenas se separan por completo. La temperatura a la que la mitad del ADN se ha convertido en una sola cadena se le llama punto de fusión, Tm.
Para medir el grado de desnaturalización se usa la absorción de luz ultravioleta. Se hacen mediciones a una longitud de onda de 260 nm, cercana al máximo de absorbencia para los ácidos nucleicos. El ADN de una hebra absorbe 12 a 14% más luz que el ADN de doble hebra a 260 nm. Una gráfica del cambio de absorbencia de una solución de ADN en función de la temperatura se llama curva de fusión. La absorbencia aumenta en forma marcada en el punto de fusión, y la transición de ADN de doble hebra a una hebra se efectúa dentro de límites estrechos de temperatura.
 |
| curva de fusión |
D. Conformación de ADN de doble hebra
El ADN de doble hebra puede asumir distintas conformaciones bajo condiciones diferentes. La conformación local también se afecta por dobleces en la molécula de ADN, y de si está unida a una proteína. El resultado es que la cantidad de pares de bases por
vuelta en el B-ADN puede fluctuar de 10.2 a 10.6.Además de varias formas de B-ADN, hay dos conformaciones muy diferentes del ADN. Se forma A-ADN cuando se deshidrata el ADN, y se puede formar Z-ADN cuando están presentes ciertas secuencias. (Las formas A y B de ADN fueron descubiertas por Rosalind Franklin en 1952). El A-ADN está enrollado más apretadamente que el B-ADN, y los surcos mayor y menor del A-ADN tienen ancho similar. Hay unos 11 pb por vuelta en el A-ADN, y los pares de bases están desplazados unos 20° en relación al eje mayor de la hélice. El Z-ADN difiere todavía más del B-ADN. En el ZADN no hay surcos y la hélice es izquierda, no derecha. La conformación Z-ADN está en regiones ricas en G/C. Los residuos de desoxiguanilato en el Z-ADN tienen distinta conformación de azúcares (3 -endo) y la base tiene la conformación sin. Tanto las conformaciones A-ADN como Z-ADN existen in vivo, pero se confinan a cortas regiones del ADN.

Superenrollamiento del ADN
 Una molécula circular de ADN con la conformación B tiene un promedio de 10.4 pares de bases por vuelta. Se dice que está relajada si tal molécula puede reposar plana sobre una superficie. Esta doble hélice relajada se puede seguir envolviendo o desenvolviendo si se rompen las hebras del ADN y se tuercen los dos extremos de la molécula lineal en direcciones opuestas. Cuando se vuelven a unir las hebras para crear un círculo, ya no hay 10.4 pares de bases por vuelta, las necesarias para mantener la conformación B estable. La molécula circular se compensa por el envolvimiento o desenvolvimiento formando superenrollamientos que restauran 10.4 pares de bases por vuelta de la doble hélice. Cada superenrollamiento se compensa por una vuelta de la doble hélice.
Una molécula circular de ADN con la conformación B tiene un promedio de 10.4 pares de bases por vuelta. Se dice que está relajada si tal molécula puede reposar plana sobre una superficie. Esta doble hélice relajada se puede seguir envolviendo o desenvolviendo si se rompen las hebras del ADN y se tuercen los dos extremos de la molécula lineal en direcciones opuestas. Cuando se vuelven a unir las hebras para crear un círculo, ya no hay 10.4 pares de bases por vuelta, las necesarias para mantener la conformación B estable. La molécula circular se compensa por el envolvimiento o desenvolvimiento formando superenrollamientos que restauran 10.4 pares de bases por vuelta de la doble hélice. Cada superenrollamiento se compensa por una vuelta de la doble hélice.
La mayor parte de las moléculas de ADN circular están superenrrolladas en las células, pero hasta las moléculas lineales largas de ADN contienen regiones localmente sobretorcidas. Los cromosomas bacterianos, en forma típica, tienen unos cinco superenrollamientos por 1 000 pares de bases de ADN. Según se verá en la sección 19.5, el ADN en los núcleos de las células eucarióticas también está sobretorcido. Todos los organismos tienen enzimas que pueden romper al ADN, destorcer o sobretorcer la doble hélice y volver a unir las hebras para alterar la topología. Esas enzimas se llaman topoisomerasas, y se encargan de agregar y eliminar superenrollamientos.
 |
| Topoisomerasas I humana |
La información contenida en el genoma debe especificar la estructura primarias de cada proteína en un organismo.
- Gen: secuencia de ADN que se transcribe a ARN. Esta definición también engloba los genes que no codifican proteínas.
- Genes domésticos: que codifican proteínas o moléculas de ARN que son esenciales para las actividades en todas las células vivas, (Ej. Enzimas que intervienen en procesos metabólicos).
- Genes especiales: que solo se transcriben en circunstancias especiales, (Ej. Durante la división celular) o genes que solo se expresen en un cierto tipo de células (Ej. la insulina solo se produce en las células pancreáticas).
- La cantidad de genes van desde 15 000 en Drosophila melanogaster a mas de 50 000 en mamíferos.
Diversos tipos de ARN en las células
Las moléculas de ARN participan en varios procesos asociados a la expresión génica. Esas moléculas se encuentran en copias múltiples y en varias formas distintas dentro de una célula dada. Hay cuatro clases principales de ARN en todas las células vivas:1. ARN ribosómico (ARNr): moléculas que son parte integral de los ribosomas (ribonucleoproteínas intracelulares que son sitios de síntesis de proteínas). El ARNribosómico es la clase más abundante de ácido ribonucleico, que forma 80% delARN celular total.
2. ARN de transferencia (ARNt): son moléculas que llevan a los aminoácidos activados a los ribosomas para su incorporación a las cadenas de péptidos en crecimiento durante la síntesis de proteínas. Las moléculas de ARNt sólo tienen de 73 a 95 residuos de nucleótidos de longitud. Forman un 15% del ARN celular total.
3. ARN mensajero(ARNm): moléculas que codifican las secuencias de aminoácidos en las proteínas. Son los “mensajeros” que llevan la información del ADN al complejo de traducción, donde se sintetizan las proteínas. En general, el ARNm sólo forma el 3% del ARN celular total. Estas moléculas son las menos estables de los ácidos ribonucleicos celulares.
4. ARN pequeño: moléculas presentes en todas las células. Algunas moléculas pequeñas de ARN tienen actividad catalítica o contribuyen a la actividad catalítica, asociadas a proteínas. Muchas de esas moléculas de ARN se relacionan con eventos de procesamiento que modifican al ARN después de que se ha sintetizado.
Las moléculas de ARN participan en varios procesos asociados a la expresión génica. Esas moléculas se encuentran en copias múltiples y en varias formas distintas dentro de una célula dada. Hay cuatro clases principales de ARN en todas las células vivas:1. ARN ribosómico (ARNr): moléculas que son parte integral de los ribosomas (ribonucleoproteínas intracelulares que son sitios de síntesis de proteínas). El ARNribosómico es la clase más abundante de ácido ribonucleico, que forma 80% delARN celular total.
2. ARN de transferencia (ARNt): son moléculas que llevan a los aminoácidos activados a los ribosomas para su incorporación a las cadenas de péptidos en crecimiento durante la síntesis de proteínas. Las moléculas de ARNt sólo tienen de 73 a 95 residuos de nucleótidos de longitud. Forman un 15% del ARN celular total.
3. ARN mensajero(ARNm): moléculas que codifican las secuencias de aminoácidos en las proteínas. Son los “mensajeros” que llevan la información del ADN al complejo de traducción, donde se sintetizan las proteínas. En general, el ARNm sólo forma el 3% del ARN celular total. Estas moléculas son las menos estables de los ácidos ribonucleicos celulares.
4. ARN pequeño: moléculas presentes en todas las células. Algunas moléculas pequeñas de ARN tienen actividad catalítica o contribuyen a la actividad catalítica, asociadas a proteínas. Muchas de esas moléculas de ARN se relacionan con eventos de procesamiento que modifican al ARN después de que se ha sintetizado.
ARN polimerasa
La ARN polimerasa es una ARN nucleotidiltransferasa. Su función es llevar a cabo la transcripción. Realiza una copia de ADN a ARN catalizando la formación de los enlaces fosfodiester entre ribonucleótidos. La copia la hace nucleótido a nucleótido, usando ribonucleósidos trifosfato (rNTP). En el ARN el ribonucleótido uracilo sustituye a la timina del ADN.La ARN polimerasa en general es oligomérica, estructurándose en un complejo formado por varias subunidades. Las subunidades comunes más grandes son dos: la β, que funciona como centro activo; y la β', que es la subunidad de unión al ADN. Está formado por una larga cola de 52 repeticiones del heptapéptido “YSPTSPS” con residuos fosforilables. El resto de subunidades de la ARN polimerasa son más pequeñas y participan en la unión a todos las proteínas que intervienen en la transcripción.
Las principales ARN polimerasas de eucariotas son la I, la II, la III, y la mitocondrial
Las principales ARN polimerasas de eucariotas son la I, la II, la III, y la mitocondrial
Los diferentes tipos de ARN polimerasas se diferencian en su composición de
aminoácidos, en su estructura, en su localización, en el tipo de ARN que
transcriben y en su forma de inhibición.
ARN polimerasa I: síntesis, reparación y revisión. Sintetiza precursores de ARN ribosómico.
ARN polimerasa II: reparación, sintetiza precursores de ARN mensajero, microARN y otros tipos de ácido ribonucleico. Esta polimerasa es el tipo más estudiado, y se requieren factores de transcripción para que se una a los promotores del ADN.
ARN polimerasa III: sintetiza ARN de transferencia, ARN ribosómico de 5S y otros pequeños ARN (ARNpequeños) encontrados en el núcleo celular (ARNp nucleares) y en el citoplasma (ARNp citoplasmáticos).
Otros tipos de ARN polimerasa se encuentran en la mitocondria y en cloroplasto
y en el núcleo del ribosoma.
aminoácidos, en su estructura, en su localización, en el tipo de ARN que
transcriben y en su forma de inhibición.
ARN polimerasa I: síntesis, reparación y revisión. Sintetiza precursores de ARN ribosómico.
ARN polimerasa II: reparación, sintetiza precursores de ARN mensajero, microARN y otros tipos de ácido ribonucleico. Esta polimerasa es el tipo más estudiado, y se requieren factores de transcripción para que se una a los promotores del ADN.
ARN polimerasa III: sintetiza ARN de transferencia, ARN ribosómico de 5S y otros pequeños ARN (ARNpequeños) encontrados en el núcleo celular (ARNp nucleares) y en el citoplasma (ARNp citoplasmáticos).
Otros tipos de ARN polimerasa se encuentran en la mitocondria y en cloroplasto
y en el núcleo del ribosoma.
Reacción catalizada por la ARN polimerasa
Cuando un ribonucleosido trifosfato entrante se aparea en forma correcta con el siguiente nucleótido no se aparea en la hebra de plantilla de ADN, la ARN polimerasa cataliza un ataque nucleofilico del grupo 3’-hidroxilo de la hebra creciente de ARN, al átomo de fosforo α- del ribonucleosido trifosfato entrante. El resultado es que se forma un fosfodiester y se libera un pirofosfato. La hidrolisis siguiente del pirofosfato catalizada por la pirofosfatasa suministra una fuerza impulsora termodinámica adicional para la reacción.
Cuando un ribonucleosido trifosfato entrante se aparea en forma correcta con el siguiente nucleótido no se aparea en la hebra de plantilla de ADN, la ARN polimerasa cataliza un ataque nucleofilico del grupo 3’-hidroxilo de la hebra creciente de ARN, al átomo de fosforo α- del ribonucleosido trifosfato entrante. El resultado es que se forma un fosfodiester y se libera un pirofosfato. La hidrolisis siguiente del pirofosfato catalizada por la pirofosfatasa suministra una fuerza impulsora termodinámica adicional para la reacción.
Comentarios de los vídeos:
Replicación del ADN: en este vídeo se presenta el proceso de replicación de ADN, en la cual mecanismo que permite al ADN duplicarse (es decir, sintetizar una copia idéntica). De esta manera de una molécula de ADN única, se obtienen dos o más "réplicas" de la primera.Esta duplicación del material genético se produce de acuerdo con un mecanismo semiconservador,lo que indica que los dos polímeros complementarias del ADN original, al separarse, sirven de molde cada una para la síntesis de una nueva cadena complementaria de la cadena molde, de forma que cada nueva doble hélice contiene una de las cadenas del ADN original. La molécula de ADN se abre como una cremallera por ruptura de los puentes de hidrógeno entre las bases complementarias puntos determinados: los orígenes de replicación. Las proteínas iniciadoras reconocen secuencias de nucleótidos específicas en esos puntos y facilitan la fijación de otras proteínas que permitirán la separación de las dos hebras de ADN formándose una horquilla de replicación. Un gran número de enzimas y proteínas intervienen en el mecanismo molecular de la replicación, formando el llamado complejo de replicación
Transcripción de ADN A ARN: La transcripción del ADN es el primer proceso de la expresión génica, mediante el cual se transfiere la información contenida en la secuencia del ADN hacia la secuencia de proteína utilizando diversos ARN como intermediarios. Durante la transcripción genética, las secuencias de ADN son copiadas a ARN mediante una enzima llamada ARN polimerasa que sintetiza un ARN mensajero que mantiene la información de la secuencia del ADN. De esta manera, la transcripción del ADN también podría llamarse síntesis del ARN mensajero.
Traducción (de ADN A proteína): La traducción es el segundo proceso de la síntesis proteica (parte del proceso general de la expresión génica). La traducción ocurre tanto en el citoplasma, donde se encuentran los ribosomas, como también en el retículo endoplasmático rugoso (RER). Los ribosomas están formados por una subunidad pequeña y una grande que rodean al ARN. En la traducción, el ARN mensajero se decodifica para producir un polipéptido específico de acuerdo con las reglas especificadas por el código genético. Es el proceso que convierte una secuencia de ARN mensajero en una cadena de aminoácidos para formar una proteína. Es necesario que la traducción venga precedida de un proceso de transcripción. El proceso de traducción tiene tres fases: iniciación, elongación y terminación (entre todos describen el crecimiento de la cadena de aminoácidos, o polipéptido, que es el producto de la traducción).
Medicamentos que contienen Ácidos nucleíco
Abacavir
Es un fármaco sintético análogo de los nucleósidos, inhibidor de la transcriptasa inversa, que es utilizado en el tratamiento contra el VIH, causante del sida. Existe una marca comercial que se expende en combinación con otros fármacos antivirales (abacavir, zidovudina y lamivudina). Fue aprobado para uso público en 1998.

Indicaciones
 El fármaco es usado para tratar el HIV tipo 1 y debe siempre ser utilizado en combinación con otros agentes antirretrovirales. Abacavir jamás debe usarse como único fármaco cuando se cambien los regímenes antirretrovirales debido a pérdida de la respuesta viral.
El fármaco es usado para tratar el HIV tipo 1 y debe siempre ser utilizado en combinación con otros agentes antirretrovirales. Abacavir jamás debe usarse como único fármaco cuando se cambien los regímenes antirretrovirales debido a pérdida de la respuesta viral.
Mecanismo de acción
El abacavir es un análogo de la guanosina (una purina). Su objetivo es la inhibición de la enzima transcriptasa inversa.
Dosis
Las propiedades farmacocinéticas del abacavir han sido estudiadas en pacientes adultos asintomáticos infectados de HIV después de una dosis única i.v. de 150 mg y de dosis orales múltiples. Las constantes farmacocinéticas del abacavir fueron independientes de la dosis entre 300 y 1200 mg/día.
Nevirapina:
 La nevirapina (BI-RG-587) es un inhibidor de la transcriptasa inversa no nucleósido (INNTI) con actividad frente al VIH-1. Estructuralmente es un miembro del grupo de las dipiridodiazepinonas (C15H14N4O; PM 266.3). La nevirapina actúa por un mecanismo de inhibición no competitiva reversible (1). Se une directamente a la trascriptasa inversa (TI) causando una alteración del lugar catalítico del enzima (2) y bloqueando las actividades de la ADN polimerasa, ARN-dependientes y ADN-dependientes.
La nevirapina (BI-RG-587) es un inhibidor de la transcriptasa inversa no nucleósido (INNTI) con actividad frente al VIH-1. Estructuralmente es un miembro del grupo de las dipiridodiazepinonas (C15H14N4O; PM 266.3). La nevirapina actúa por un mecanismo de inhibición no competitiva reversible (1). Se une directamente a la trascriptasa inversa (TI) causando una alteración del lugar catalítico del enzima (2) y bloqueando las actividades de la ADN polimerasa, ARN-dependientes y ADN-dependientes. Dosis:
Dosis:
Dosis adulto: 200 mg/día durante 14 días, aumentando posteriormente a 200 mg/12 h oral, con o sin comida (esta pauta de inicio ha reducido la incidencia de exantema).
Dosis niño: 120 mg/m2/24 h durante 28 días, seguido de 200 mg/m2/12 h en niños <9 años y de 120 mg/m2/12 h en niños >9 años.
Aciclovir

Es un fármaco antiviral que se usa en el tratamiento de las infecciones producidas por la varicela, el virus herpes humano (VHH), entre las que se incluyen el herpes bucal, el herpes zóster y la mononucleosis infecciosa.
Este fármaco impide la replicación viral disminuyendo la extensión y duración de la enfermedad.
Vías de administración
El aciclovir se usa principalmente por vía oral mediante formulaciones en comprimidos y suspensión para el uso pediátrico. También se administra por vía tópica, en crema, e intravenoso en pacientes con infecciones graves por herpes virus.
Mecanismo de Acción
En su forma absorbible, el aciclovir tiene poca afinidad a las enzimas de células no infectadas. Es convertido selectivamente en una forma monofosfatada por una timidina quinasa que poseen los virus sensibles al medicamento. Subsecuentemente, el monofosfato es fosforilado para ser convertido, primero en difosfato - aciclo-GDP - y luego en el trifosfato activo - aciclo GTP, por quinasas celulares.
 El aciclo-GTP inhibe la síntesis de ADN viral a través de un mecanismo competitivo con la polimerasa viral, y al ser incorporada en la cadena de ADN en síntesis, detiene su replicación. Su poca afinidad a las polimerasas celulares, sumado al hecho de que la fosforilación ocurre sólo en células infectadas, hace que tenga una toxicidad baja.
El aciclo-GTP inhibe la síntesis de ADN viral a través de un mecanismo competitivo con la polimerasa viral, y al ser incorporada en la cadena de ADN en síntesis, detiene su replicación. Su poca afinidad a las polimerasas celulares, sumado al hecho de que la fosforilación ocurre sólo en células infectadas, hace que tenga una toxicidad baja.
Dosis
Para adultos y niños mayores de 12 años, la vía de administración es la infusión intravenosa de 5 mg/kg de peso cada 8 h, durante cinco días. La solución se preparara como indica el fabricante y debe administrarse lentamente durante por lo menos 1 h. Para el herpes simple y herpes zóster diseminado, se administra como se indica durante siete días. La vía de administración oral es de 200 mg cinco veces al día, a intervalos de 4 h, durante cinco o más días.
Para Niños (menores de 12 años), la infusión intravenosa es de 250 mg/m2 de superficie corporal cada 8 h, durante cinco días. Para el herpes simple, se administra durante siete días. El medicamento está prescrito de preferencia por especialistas en casos graves y en pacientes hospitalizados. En varicela se administran 20 mg/kg/día sin pasar de 800 mg en 24 h durante por 5 días.
No hay comentarios.:
Publicar un comentario